








Association analysis of repetitive elements and R-loop formation across species
Preprint posted on 10 November 2020 https://www.biorxiv.org/content/10.1101/2020.11.09.374124v1
Article now published in Mobile DNA at https://mobilednajournal.biomedcentral.com/articles/10.1186/s13100-021-00231-5
Looped in the repeats: correlating R-loops with repetitive genetic elements.
Selected by Sree Rama ChaitanyaCategories: genomics, molecular biology
Context1-3
R-loops are non-canonical three-stranded nucleic acid structures that are formed when the RNA hybridizes with the complementary DNA strand displacing the other strand free. The factors that influence the formation and genome-wide distribution of R-loops include several proteins involved in transcription, splicing, replication, recombination, DNA repair, and chromatin modifiers, etc. Additionally, R-loops tend to form at repetitive elements and skewed sequences (GC-skew and AT-skew). However, it is not clear if R-loops have any sequence bias in their genome-wide distribution among different species. Therefore, the authors of the current preprint looked into published datasets to understand the cis-regulatory elements associated with genome-wide R-loop distribution.
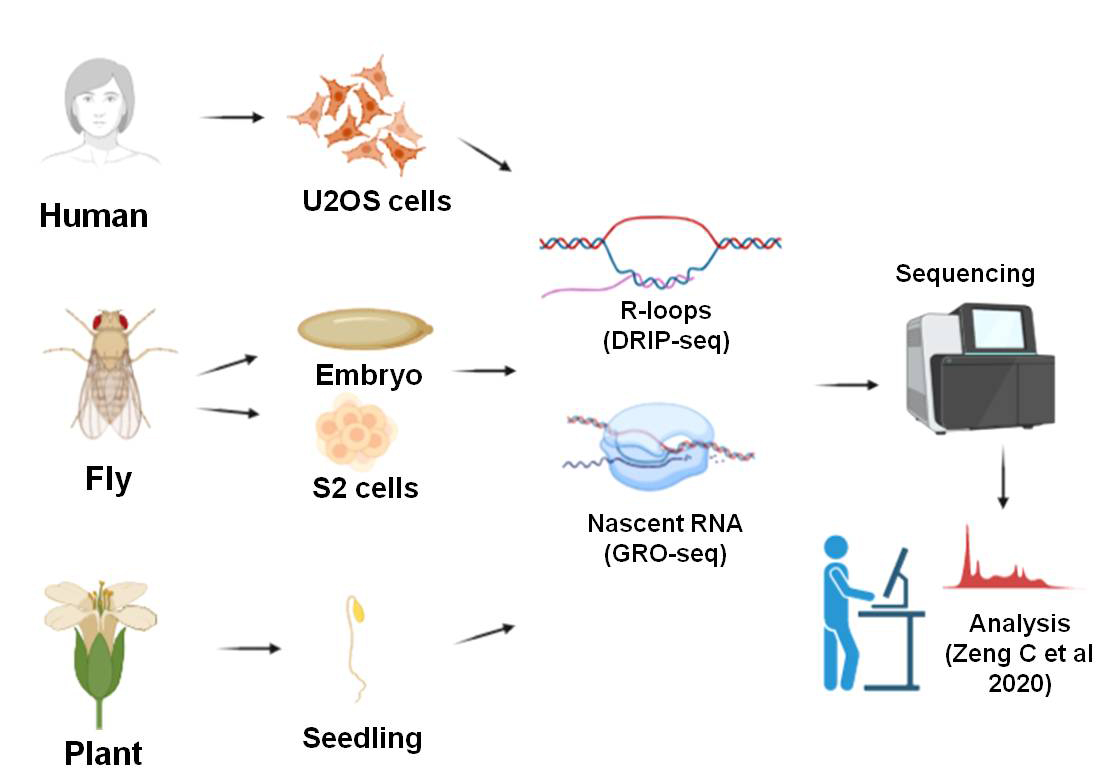
Key findings
- The authors reanalyzed publicly available datasets generated in human cells (U2OS), fly (D. melanogaster embryos, S2 cells), and plants (seedling of A. thaliana) using different controls. They used R-loop (DRIP-seq) and nascent RNA profiles (GRO-seq) for the study. They observed that R-loops in plants tend to be longer (~998 nucleotides) than humans and fly (~414-618 nucleotides). Across the species, R-loops tend to enrich at gene promoters. Of note, plants harbor about 60% of the total R-loops at their promoters (and 0.2% at their introns). They also found a 70%, 24%, 39%, and 54% overlap between R-loops and transcribing regions in humans, fly embryos, S2 cells, and plants. However, when they analyzed further, flies tend to harbor R-loops more at intergenic regions (~90%) and possibly independent of transcription, suggesting the presence of trans R-loops.
- The authors report some species-specific differences. They show that human and plant R-loops were marginally enriched at ribosomal DNA and underrepresented at short interspersed nuclear elements (SINEs). But more enriched at retrotransposons and satellite DNA. This is in contrary to the fly genome that has underrepresented R-loops at the satellite DNA (they also notice some difference in R-loop genome-wide distribution in the fly genome between embryos and S2 cells, possibly reflecting the developmental stages).
- Overall, all the species analyzes showed a positive correlation between repetitive genetic elements and R-loop genome-wide distribution. In human cells, telomeres, centromeres, ribosomal DNA, and retrotransposons are enriched for R-loops. In the fly genome, Long interspersed nuclear elements (LINEs), Long terminal repeats (LTRs), and low complexity regions enriched for R-loops. However, in the plant genome, about half of the repeat families were enriched in R-loops; these include LINEs, LTRs, and low complexity regions, etc.
Perspective
Either a cause or consequence, R-loops seem to play a crucial role in developmental pathways, cancer progression, and neurodegenerative diseases. Thus, many researchers are drawn to understand their precise physiological role. While most of the work in R-loop biology looked at trans-acting factors, here, the authors investigated the association of cis-regulatory elements or sequence determinants of R-loop formation. The authors found strong correlations between R-loops and repetitive DNA sequences reinforcing earlier studies.
(Note: I only highlighted the key findings of the preprint without commenting on the methodology. Anyone is free to comment on the methodology, in case the preprint excites you.)
Acknowledgments: I am thankful to all the authors for their support, especially Chao Zeng for taking the time to comment on the preLight.
References:
- https://doi.org/10.1093/nar/gkw1054
- https://doi.org/10.1101/gr.158436.113
- https://doi.org/10.1038/s41477-017-0004-x
- https://doi.org/10.7554/elife.17548
- https://doi.org/10.1038/s41467-017-00338-5
- https://doi.org/10.1016/j.cub.2017.01.011
Posted on: 28 December 2020 , updated on: 22 January 2021
doi: https://doi.org/10.1242/prelights.25813
Read preprint (3 votes)
(3 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Temporal constraints on enhancer usage shape the regulation of limb gene transcription
Transcriptional profiling of human brain cortex identifies novel lncRNA-mediated networks dysregulated in amyotrophic lateral sclerosis
A long non-coding RNA at the cortex locus controls adaptive colouration in butterflies
AND
The ivory lncRNA regulates seasonal color patterns in buckeye butterflies
AND
A micro-RNA drives a 100-million-year adaptive evolution of melanic patterns in butterflies and moths
Also in the molecular biology category:
Fetal brain response to maternal inflammation requires microglia
Clusters of lineage-specific genes are anchored by ZNF274 in repressive perinucleolar compartments
Nanos2+ cells give rise to germline and somatic lineages in the sea anemone Nematostella vectensis
preLists in the genomics category:
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
Also in the molecular biology category:
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |