








Conserved phosphorylation hotspots in eukaryotic protein domain families
Preprint posted on 13 August 2018 https://www.biorxiv.org/content/early/2018/08/13/391185
Article now published in Nature Communications at http://dx.doi.org/10.1038/s41467-019-09952-x
Rules of the game: extensive comparative study reveals phosphorylation hotspots in key eukaryotic protein domains
Selected by Dey LabCategories: evolutionary biology, systems biology
Context
Post-translational modifications (PTMs), and protein phosphorylation in particular, serve as the cell’s precision rewiring tools. The addition (or removal) of a single charged phosphate group can alter a protein’s enzymatic activity, drive a binding partner switch, affect folding and stability, or force a change in subcellular location- all in a matter of seconds!
Advances in mass spectrometry have made it possible to map sites of protein phosphorylation on a massive scale. In recent years, this has produced an overwhelming wealth of genome-wide data across a range of cell types and species- along with new analytical challenges. Take the human genome: of approximately 160,000 non-redundant phosphosites1, only a tiny fraction is annotated, and many might not actually be functional2, with little or no contribution to fitness. How, then, to assess the functional relevance of all this data?
Evolutionary comparisons can help, based on the argument that highly conserved phosphosites are likely to be functional. However, these comparisons are not without their own challenges. Many functional phosphosites lie within unstructured regions of proteins, presumably relaxing selective constraints on their positions. Further complicating such analyses, individual phosphosites have the capacity to flip to acidic residues (and back) on relatively short evolutionary timescales3, rewiring signaling cascades in the process4.
Building on work initiated when he was a postdoc at UCSF2, Pedro Beltrao and colleagues circumvent these challenges to generate what is, to my knowledge, the most comprehensive comparative analysis of phosphosites, encompassing more than 500,000 phosphosites across 40 eukaryotic genomes.
Major findings
The authors selected a subset 344 well-represented Pfam domain families with a high density of phosphorylation sites. Using a rolling window to account for alignment and assignment errors, and a background expectation generated by randomly permuting phosphosites to equivalent residues within the same sequence, resulted in the identification of significant “hotspots” within 162 of the 344 families. Encouragingly, the hotspots were enriched for known functional phosphosites- and once mapped onto structural models, recovered well-characterized regulatory motifs (Figure 1).
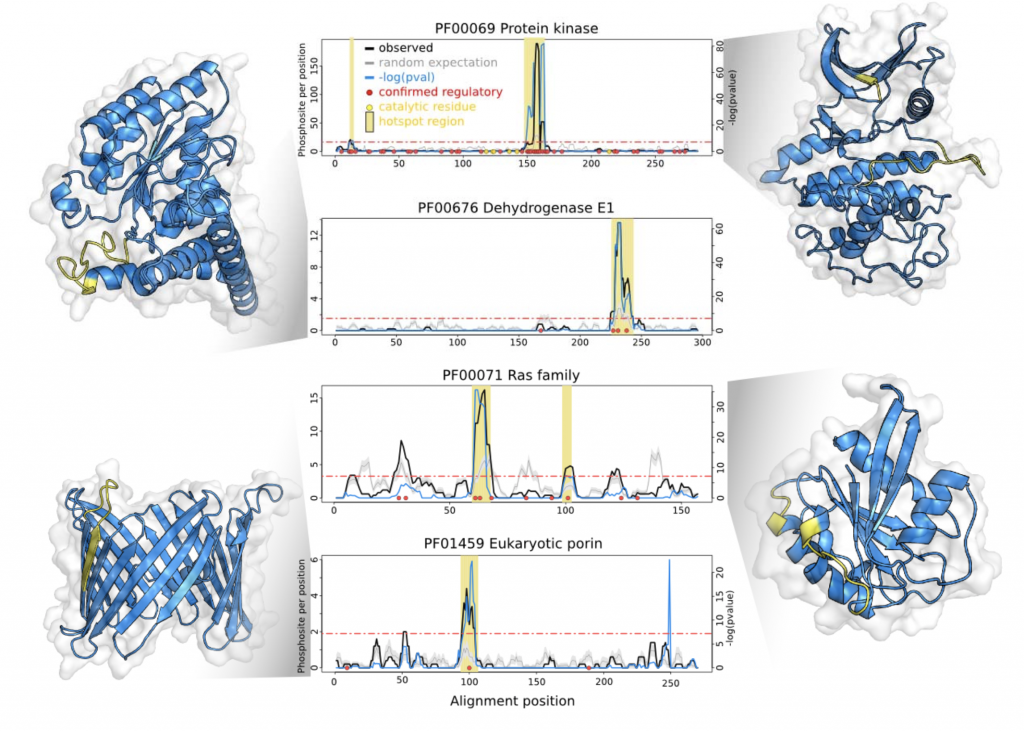
The authors then looked to generalize their analyses, and found that the hotspots tend to be located proximal to catalytic residues or binding interfaces- across a broad range of domain families. Zooming in, they make experimentally tractable functional predictions for uncharacterised hotspots in two enzymes- IMP dehydrogenase and transaldolase. Finally, they selected two phosphorylation sites within a budding yeast ribosomal S11 domain hotspot for an experimental case study of their own, demonstrating a functional role for one of them.
What’s next?
I think it’s quite clear that this study has generated a fantastic resource for the larger community, although- as the authors are quick to point out in their discussion- follow-up structural biology analyses will not necessarily be straightforward. To end with a couple of open-ended questions:
- Could the hotspot database be flipped around to help answer evolutionary questions? For example, are there any systematic differences between the organisation of hotspots in apicomplexan parasites and their free-living relatives? Or between multicellular and unicellular organisms?
- How far are we from a bottoms-up, synthetic biology approach to designing protein switches or circuits controlled by phosphorylation?
- On an even deeper evolutionary timescale: how many of the domain families are present in bacteria or archaea? It would be fascinating to extend the hotspot analysis beyond eukaryotes if feasible!
References:
- Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
- Beltrao, P. et al. Systematic Functional Prioritization of Protein Posttranslational Modifications. Cell 150, 413–425 (2012).
- Pearlman, S. M., Serber, Z. & Ferrell, J. E. A mechanism for the evolution of phosphorylation sites. Cell 147, 934–46 (2011).
- Dey, G. & Meyer, T. Phylogenetic Profiling for Probing the Modular Architecture of the Human Genome. Cell Syst. 1, 106–115 (2015).
Posted on: 16 August 2018
doi: https://doi.org/10.1242/prelights.4393
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the evolutionary biology category:
Fetal brain response to maternal inflammation requires microglia
How the liver contributes to stomach warming in the endothermic white shark Carcharodon carcharias
A long non-coding RNA at the cortex locus controls adaptive colouration in butterflies
AND
The ivory lncRNA regulates seasonal color patterns in buckeye butterflies
AND
A micro-RNA drives a 100-million-year adaptive evolution of melanic patterns in butterflies and moths
Also in the systems biology category:
Expressive modeling and fast simulation for dynamic compartments
Clusters of lineage-specific genes are anchored by ZNF274 in repressive perinucleolar compartments
Holimap: an accurate and efficient method for solving stochastic gene network dynamics
preLists in the evolutionary biology category:
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
EMBO | EMBL Symposium: The organism and its environment
This preList contains preprints discussed during the 'EMBO | EMBL Symposium: The organism and its environment', organised at EMBL Heidelberg, Germany (May 2023).
| List by | Girish Kale |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Planar Cell Polarity – PCP
This preList contains preprints about the latest findings on Planar Cell Polarity (PCP) in various model organisms at the molecular, cellular and tissue levels.
| List by | Ana Dorrego-Rivas |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
COVID-19 / SARS-CoV-2 preprints
List of important preprints dealing with the ongoing coronavirus outbreak. See http://covidpreprints.com for additional resources and timeline, and https://connect.biorxiv.org/relate/content/181 for full list of bioRxiv and medRxiv preprints on this topic
| List by | Dey Lab, Zhang-He Goh |
1
SDB 78th Annual Meeting 2019
A curation of the preprints presented at the SDB meeting in Boston, July 26-30 2019. The preList will be updated throughout the duration of the meeting.
| List by | Alex Eve |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |
Also in the systems biology category:
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |