








Deep learning-based predictions of gene perturbation effects do not yet outperform simple linear methods
Posted on: 11 November 2024 , updated on: 5 August 2025
Preprint posted on 1 October 2024
Article now published in Nature Methods at https://doi.org/10.1038/s41592-025-02772-6
Categories: bioinformatics
Updated 5 August 2025 with a postLight by Benjamin Maier
Congratulations to Constantin, Wolfgang and Simon for their manuscript now being published in Nature Methods. Geneformer, scBERT and UCE have been added to the benchmark during the revisions, yet their single- and double-perturbation predictions are also performing worse than the simple baseline models. The authors additionally contacted the authors of all the tested methods to verify that the methods were run with their optimal parameters and added some clarifications regarding test datasets, computational time and benchmark routine setup. Otherwise, the manuscript has not changed much and the conclusions from the preprint continue to hold true.
The full peer review history is available here: https://static-content.springer.com/esm/art%3A10.1038%2Fs41592-025-02772-6/MediaObjects/41592_2025_2772_MOESM2_ESM.pdf
Background
Foundation Models
Foundation models are a type of generative AI that are created mainly through a process called self-supervised learning, which uses unlabelled datasets. This means that these models can learn from vast amounts of data without the need for human intervention to label or curate it. As a result, they are much faster and more cost-effective to implement on a large scale compared to traditional supervised models that require extensive manual preparation. Furthermore, the pre-training on diverse data types (text, images, …) leads to generalised data representations allowing them to be adapted for various downstream applications, including text translation (e.g. DeepL), image analysis/classification, and content generation (such as ChatGPT and DALL-E). The two most notable classes of foundation models are OpenAI’s GTP-n class and Google’s Bidirectional Encoder Representations from Transformers (BERT) (Devlin et. al, 2018). The scale and computational demands of these models have increased massively over the past generations. For instance, GTP-4, was trained on over a trillion words and contains 1,760,000,000,000 (1.76 trillion) parameters across 120 layers at estimated training costs exceeding $100 million. This represents a tenfold increase in parameters compared to GPT-3.
Transformers
The transformer architecture is the most commonly used framework for these models, though it is not strictly required. At its core, a transformer uses a technique called self-attention (Vaswani et al., 2017), which helps the model identify and focus on the most relevant parts of the input data when making predictions. For example, when processing a sentence, the model can pay more attention to certain words that are important for understanding the overall meaning. This selective attention allows transformers to grasp complex relationships and dependencies in the data, allowing for more nuanced understanding and improved performance.
This approach marks a significant advancement over earlier neural networks architectures like multilayer perceptrons (MLPs), convolutional neural networks (CNNs) and recurrent neural networks; which are trained on task-specific data to perform a more narrow range of functions. In contrast, to the other methods, attention-based transformer models at the same time allow order preservation, can handle variable length data, have truly long-range dependencies (global context) and are parallelizable.
A great short introductory text about foundation models and transformers by Niklas Heidloff can be found here, with links for further reading. A more technical introduction about transformers can be found here.
Single and Double Perturbation Experiments
A single perturbation CRISPR experiment involves targeting a single gene at a time (either knockout, knock-in, or mutation) to assess its role and effect in a particular biological process. Similarly, double perturbation experiments modify two genes at the same time. Double perturbations are key to uncover unexpected phenotypes that go beyond simple combinations of individual perturbations, but their high combinatorial complexity (e.g. ~ 500,000,000 experiments for all gene-pair combinations) necessitates reliable computational methods to efficiently prioritise experiments and explore more conditions efficiently. If we would be able to predict double perturbation (or even higher orders) accurately, we could prioritise/narrow down the number of lab experiments, which would ultimately help our biological understanding and accelerate drug development.
Key Findings
Overview
Constantin Ahlmann-Eltze and colleagues have benchmarked the performance of two state-of-the-art foundation models, scGPT (Cui, Wang, Maan. et al., 2024) and scFoundation (Hao, Gong, Zeng. et al., 2024), as well as the popular graph-based deep learning framework GEARS (Roohani et al., 2024) against baseline linear models. Their analysis demonstrated that all three models despite high computational training/fine-tuning expenses do not perform better for single and double perturbation predictions.
Double Perturbation Prediction
The authors used single and double perturbation CRISPR data (Norman et al., 2019) to fine-tune scGPT, scFoundation and GEARS, and subsequently assess their performance of predicting unseen (held-out) double perturbation experiments. In their benchmark, the authors compared the three state-of-the-art models against two simplistic models: one that predicts unperturbed control expression (no change) and another that sums the effects of individual perturbations (additive). Their benchmark revealed that the simple additive model outperformed the state-of-the-art models as shown by the lower predictive error of the simpler additive model (Figure 1). This is even more remarkable as the additive model has never seen the double perturbation training data and does not require any computationally expensive model training/fine-tuning. The authors further showed that the prediction performance was worse for lowly abundant genes (lower signal-to-noise ratio) and that genes with a lower differential expression are more difficult to predict.
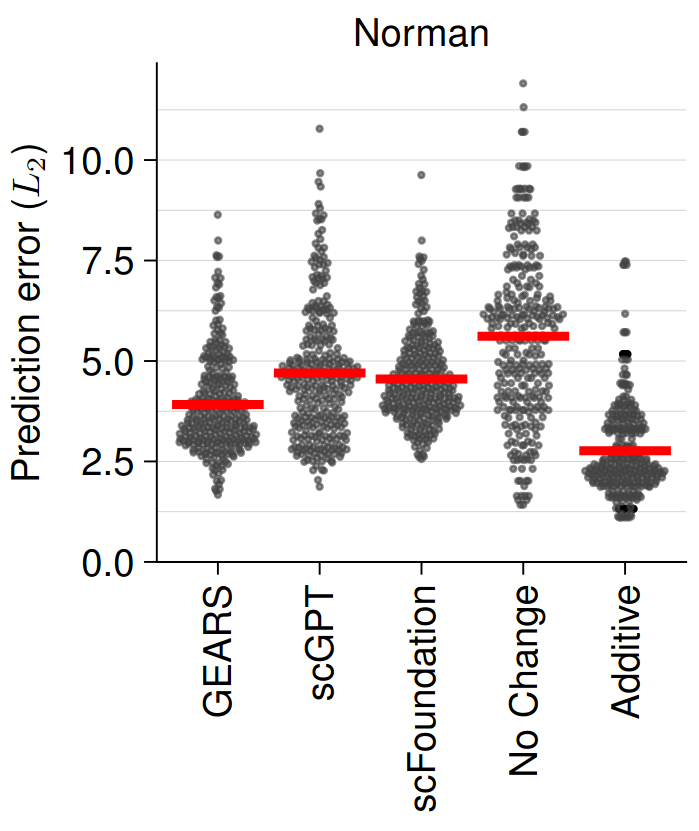
Fig. 1 Double Prediction Error (lower is better). Figure taken Figure taken from Ahlmann-Eltze et al. (2024), BioRxiv published under the CC-BY-NC 4.0 International licence.
Next, they identified 8,060 observations that they classified as “true” synergistic and repressive based on the difference between the observed logarithmic fold change and the sum of the individual ones (FDR: 5%). This was done to see whether the models might be able to explain effects that go beyond the simple additive effect of single perturbations. However, when predicting synergistic interactions, the no change model simply predicting the unperturbed state outperformed the other methods in all metrics (FDR, ROC, recall). When looking at which genes caused synergistic effects, they found that only a small number of genes contributed to most of the top synergistic interactions across all methods.
Single Perturbation Prediction
In addition to predicting unseen double perturbation experiments, foundation models can also predict the effect of unseen single perturbations relying on biological relationships learned during training or, in the case of GEARS, shared gene ontology annotations. Using three publicly available CRISPR interference dataset (Adamson et al., 2016 & Replogle et al., 2022), the authors benchmarked the single perturbation experiment prediction performance of scGPT and GEARS, which again did not outperform a simple linear baseline model (Figure 2).
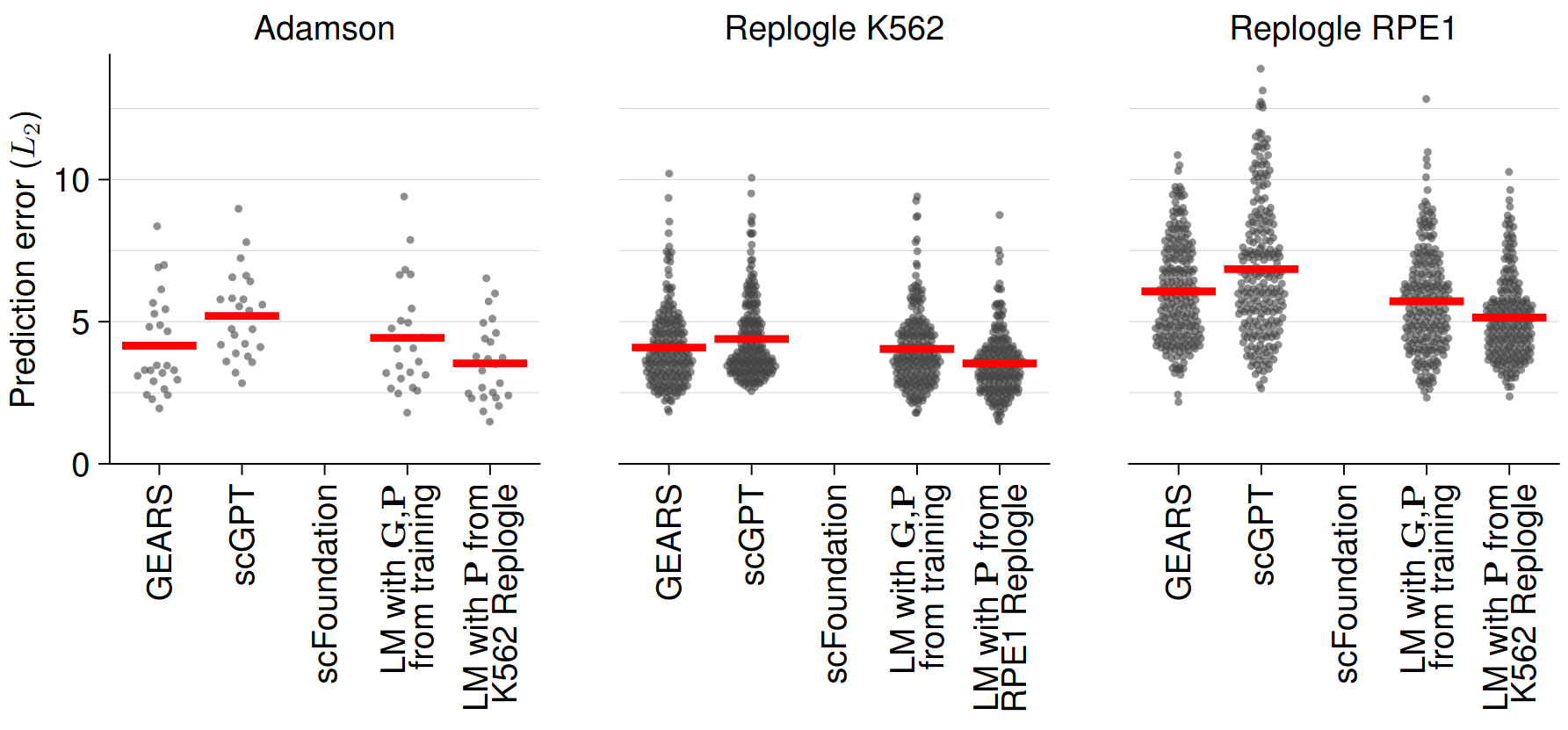
Fig. 2 Single Perturbation Prediction Error. Figure taken from Ahlmann-Eltze et al. (2024), BioRxiv published under the CC-BY-NC 4.0 International licence.
Transfer Learning Meets Foundation Models
As the linear model from the previous section was not allowed to see the test perturbations in the training phase, the authors constructed the perturbation embeddings from the gene embeddings and not the actual perturbations. Wondering whether the pre-trained perturbation embeddings from one dataset could help to improve the prediction performance of another dataset, they used the PCA from one dataset and could see an improvement in prediction performance. Next, they looked at the embeddings learnt by the state-of-the-art methods and found that they can further improve the results thereby demonstrating a proof-of-principle of transfer learning here.
Conclusion and Perspective
This study by Constantin Ahlmann-Eltze and colleagues as well as numerous other recent benchmarking papers have demonstrated that foundation models are not yet outperforming simpler models for various bioinformatics tasks such as single-cell data clustering or perturbation predictions. However, their study also revealed that the embeddings learned by the foundation models hold value independently of the transformer module. If you want to check out all the presented analysis in the manuscript and reproduce them, their code is deposited at GitHub and all used datasets are publicly available.
Personally, I really enjoyed the article for two main reasons: (1) it is an important negative result and making it available will prevent researchers from incorrectly interpreting their data as well as stimulating others to think about how to improve the methodology (see Twitter discussions surrounding this article), and (2) it showcases the value of embeddings even without the transformer module. Looking ahead, I anticipate that neural networks and AI-based methods will soon surpass simpler approaches, especially with the exponential increase in experimental data. At the same time, I believe that we must be careful not to implement AI methods for every question merely to capitalise on the AI trend, since traditional methods will still remain more suitable for many applications.
Conflict of interest
The research featured in this preLights post was conducted at the European Molecular Biology Laboratory (EMBL), where the author of this highlight post is currently enrolled in its international PhD program.
References
Adamson, B., Norman, T. M., Jost, M., Cho, M. Y., Nuñez, J. K., Chen, Y., … Weissman, J. S. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell, 167(7), 1867-1882.e21. https://doi.org/10.1016/j.cell.2016.11.048
Cui, H., Wang, C., Maan, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat Methods 21, 1470–1480 (2024). https://doi.org/10.1038/s41592-024-02201-0
Devlin, J., Chang, M.-W., Lee, K., & Toutanova, K. (2018). BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. CoRR, abs/1810.04805. arxiv, https://doi.org/10.48550/arXiv.1810.04805
Hao, M., Gong, J., Zeng, X. et al. Large-scale foundation model on single-cell transcriptomics. Nat Methods 21, 1481–1491 (2024). https://doi.org/10.1038/s41592-024-02305-7
Norman, T. M., Horlbeck, M. A., Replogle, J. M., Ge, A. Y., Xu, A., Jost, M., Gilbert, L. A., & Weissman, J. S. (2019). Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science (New York, N.Y.), 365(6455), 786–793. https://doi.org/10.1126/science.aax4438
Replogle, J. M., Saunders, R. A., Pogson, A. N., Hussmann, J. A., Lenail, A., Guna, A., Mascibroda, L., Wagner, E. J., Adelman, K., Lithwick-Yanai, G., Iremadze, N., Oberstrass, F., Lipson, D., Bonnar, J. L., Jost, M., Norman, T. M., & Weissman, J. S. (2022). Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell, 185(14), 2559–2575.e28. https://doi.org/10.1016/j.cell.2022.05.013
Roohani, Y., Huang, K. & Leskovec, J. Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nat Biotechnol 42, 927–935 (2024). https://doi.org/10.1038/s41587-023-01905-6
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., … Polosukhin, I. (2017). Attention Is All You Need. arXiv [Cs.CL]. arxiv, https://doi.org/10.48550/arXiv.1706.03762
doi: https://doi.org/10.1242/prelights.38664
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |