








Depression of heart rate in fish at critically high temperatures is due to atrioventricular block
Posted on: 26 March 2020
Preprint posted on 18 March 2020
Article now published in The Journal of Experimental Biology at http://dx.doi.org/10.1242/jeb.225227
Fish heart electrical excitation fails at high temperature due to ion current mismatch resulting in atrioventricular block.
Selected by James MarchantBackground
Contraction of the heart is initiated by electrical excitation which originates in pacemaker cells situated at the base of the heart. Propagation of the electrical signal from the sinoatrial node causes contraction of the atrium followed by the ventricle causing blood to be ejected from the ventricle into the bulbus arteriosus. The propagation of the electrical signal is orchestrated by opening and closing of voltage-gated ion channels which propagate the signal from cell to cell and chamber to chamber, passing from the atrium to the ventricle via the atrioventricular node. At critically high temperature, however, there is a breakdown of cardiac contraction which could originate in either the mechanism of electrical initiation or propagation. This article explores the latter and reveals the imbalance of rectifying K+ channels and depolarizing Na+ channels in the rainbow trout heart leads to atrioventricular block and loss of ventricular contraction in the presence of an atrial excitatory P wave.
Key findings
The temperature-dependent depression of cardiac activity was recorded via implanted ECG probes in rainbow trout (Oncorhynchus mykiss). ECG recordings revealed (2:1) atrioventricular block at high temperature (»25°C), manifesting as a missing QRS complex, which transformed into 3:1 atrioventricular block with two successive missing QRS complexes in the presence of a maintained P wave (Figure 1. A-E). As temperature is increased, heart rate increased, PQ and QT intervals decreased. However, ventricular heart rate was lower than articular at critically high temperatures, due to the development of atrioventricular block. Interestingly, the QRS duration is prolonged as seen in type 2 AV block, suggesting failure of the ventricular conduction system.
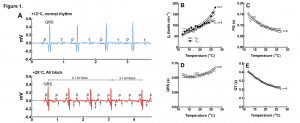
Figure 1. Effects of acute warming on ECG of rainbow trout. (A) Representative ECG recordings at +12°C and +25°C. The lower panel shows 2:1 atrioventricular block which ends up in 3:1 AV block (successive missing of two QRS complexes, small arrows). (B) Effect of temperature on atrial (fHA; n=4) and ventricular (fHV; n=8) beating rate determined from the number of P waves and QRS complexes, respectively. (C-E) Effect of temperature on PQ interval (C), QRS duration (D) and QT interval (E). Results are means of 8 fish. Dotted lines show ±95% confidence limits.
The authors then looked at the cellular effects of high temperature in isolated ventricular myocytes and found that the resting membrane potential and the voltage-gated Na+ channel threshold potential had both become more negative, but that the change in resting membrane potential was greater, increasing critical depolarization required for reaching threshold potential. (Figure 2. A-F). Action potential amplitude was also increased at high temperature (25°C) whereas action potential duration 50 decreased and both overshoot potential and the maximum rates of AP depolarization (+ dV/dt) and repolarization (-dV/dt) remained the same.
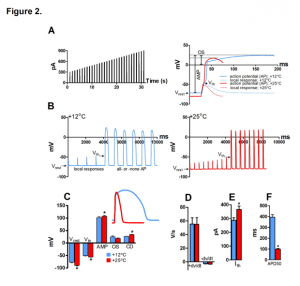
Figure 2. Effect of temperature on AP initiation of rainbow trout ventricular myocytes at +12°C and +25°C. (A) Stimulus current protocol (left) and representative fast sweep recordings of Aps at +12°C and +25°C. The measured AP parameters are shown for the AP at +12°C (right). (B) Slow sweep recordings of ventricular APs +12°C (left) and +25°C (right) showing smaller voltage responses to stimulus current and a more negative Vth at +25°C compared at +12°C (left). (C) Effects of temperature resting membrane potential (Vrest), threshold potential (Vth), AP amplitude (AMP), AP overshoot (OS) and critical depolarization (CD). (D) Effects of temperature on the maximum rates of AP depolarization (+ dV/dt) and repolarization (-dV/dt). Effects of temperature on threshold current (Ith) (E) and AP duration at 50% of repolarization (APD50) (F).
Also in isolated ventricular cardiomyocytes, the authors looked at input resistance (Rin), which is a measure of membrane leakiness. Rin was reduced more than 2-fold at 25°C compared to 12°C (528 ± 59 MΩ vs 253 ± 28 MΩ) indicating greater membrane leak at warmer temperatures resulting in increased charge requirement to trigger an action potential (Figure 3. A-B). Consequently, critical depolarization was significantly increased, as was the threshold current indicating reduced electrical excitation of ventricular myocytes at high temperature.
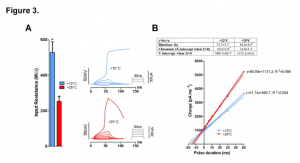
Figure 3. Effect of temperature on input resistance (Rin) and strength-duration relationship of rainbow trout ventricular myocytes. (A) Rin of resting ventricular myocytes at +12°C and +25°C. (B) Effect of temperature on strength-duration relationship of ventricular myocytes. Representative current-clamp recordings showing the requirement of increased stimulus current/charge for AP initiation of the same myocyte at +25°C relative to that at +12°C (left). Mean results of strength-duration relationship at +12°C and +25°C presented in the form of Weiss plots (right) (n=13 myocytes from 4 fishes). The dotted lines indicate ± 95% confidence charge for infinitely short pulse (y intercepts of the lines) are shown in the box above the plot.
Finally, the authors looked specifically at the sodium current (INa) and the inward rectifier current (IK1) by patch-clamp experiments. The current-voltage relationship was determined for INa at 12 and 25°C and peak density was found to be 20% larger at 25°C (-70 ± 4 pA pF-1 vs. -56 ± 3 pA pF-1 at 12°C) but charge transfer was reduced by 31% (-40 ±2 pA ms-1 pF-1) at +25°C vs. (-58 ± 3 pA ms-1 pF-1) at +12°C (Figure 4. A-C). Charge transfer of INa started to decline immediately upon warming and reached its breakpoint temperature at 18.3 ± 0.6°C, whereas IK1 breakpoint temperature was 34.0 ± 1.1°C. As the charge transfer of INadeclines and the demand for charge increases due to diverging opposing currents INa and IK1, the temperature-dependent depression of electrical excitation becomes inevitable. This was previously hypothesized by Vornanen (2016) and termed the temperature-dependent depression of electrical excitability (TDEE).
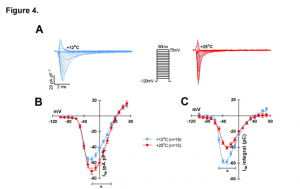
Figure 4. Effects of temperature on INa and IK1 of rainbow trout ventricular myocytes. (A) representative tracings of the voltage-dependence of INa at +12°C and +25°C. (B) Voltage- dependence of INa density at +12°C (n=19 cells from 5 fish) and +25°C (n=15 cells from 5 fish). (C) Voltage-dependence of INa charge transfer at +12°C (n=19 cells from 5 fish) and +25°C (n=15 cells from 5 fish).
In conclusion, temperature-induced decay of ventricular excitation and contractility is due to atrioventricular block caused by a mismatch between depolarizing INa current and rectifier IK1 current. Increased excitation threshold due to increased membrane leak and increased threshold potential for depolarizing INa cause an increase in critical depolarization which is not met by the depolarizing stimulus, resulting in the non-propagation of the electrical signal, causing AV block.
Why I liked this preprint
This preprint provides a simple and elegant explanation for temperature-induced depression of electrical excitation in the fish heart. The TDEE hypothesis provides a mechanistic explanation for the in vivo depression of cardiac output in fish at critically high temperature.
Questions
Charge transfer of INa is reduced at high temperature. Do you think that acclimation could counter this effect allowing the fish to maintain charge transfer at high temperature?
Is the AV block caused by interruption of impulse propagation in the AV node or due to failure of ventricular tissue to relay the excitation stimulus?
doi: https://doi.org/10.1242/prelights.17839
Read preprint (1 votes)
(1 votes)