








Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification
Posted on: 5 March 2019
Preprint posted on 1 March 2019
Article now published in Nucleic Acids Research at http://dx.doi.org/10.1093/nar/gkaa539
Targeting prokaryotic translational machinery for fragment-based drug discovery
Selected by Yasmin LauBackground
In the world of drug discovery, ongoing efforts have been made to design effective antibacterial drugs that fulfil both efficacy and specificity requirements. One such case is the development of drugs targeting translational machinery in bacteria by identifying components characteristic to their catalytic site and elucidating structure-function relationships. TrmD is an enzyme that catalyzes the methylation of a G37 base in prokaryotic tRNA which acts to reduce translational frame-shift errors (1).
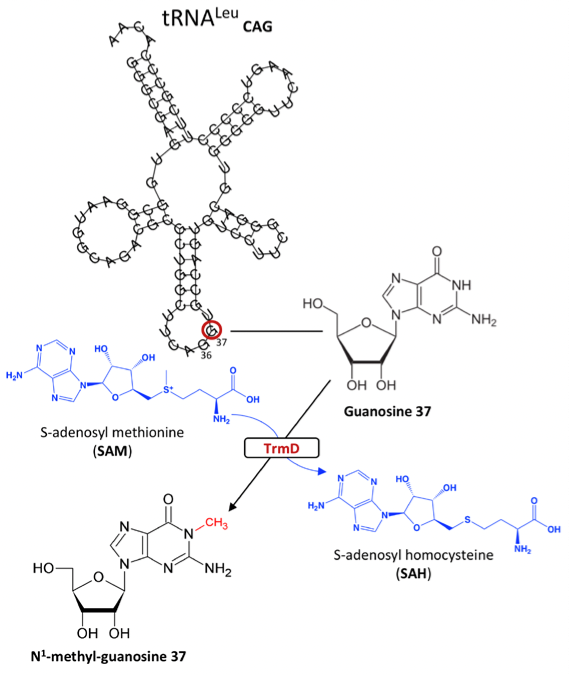
Figure 1: S-adenosyl-L-methionine-dependent methylation of Guanosine 37 via TrmD in prokaryotic tRNA
TrmD belongs to a class of S-adenosyl-L-methionine (SAM) dependent family of proteins which contain a canonical trefoil knot motif in their catalytic site. The understanding of the mechanism of action for Trm5, the enzyme responsible for G37 methylation in eukaryotes, has also been scrutinised for antibacterial drug design. However, TrmD and Trm5 differ in specificity for tRNA base G37 recognition such that the former specifically targets the D stem and anticodon stem loop of tRNA with G36G37 bases, while the latter only recognizes the L-shaped tertiary of tRNA with a G37 base. Moreover, while Trm5 binds to SAM as a monomer, TrmD binds through its trefoil knot motif and functions as a dimer. Concomitantly, by discerning enzymes attributed to certain classes, these binding and structural features can be exploited in the development of translational inhibitors. In this preprint, the authors use TrmD as a model to introduce fragment-based drug discovery, an interesting approach in which initial stages involve reducing molecular complexities to produce high ligand efficient but low potency fragments. These fragments are subsequently optimized for more efficient protein binding (2)(3).
Key findings
Determination of the structure and binding of M. abscessus TrmD
Firstly, the crystal structures of TrmD were determined, focusing on its protomers which contain a largely disordered interdomain linker (Figure 2). The trefoil knot motif which is located in the N-terminal domain is also deciphered; this is the region that binds SAM (Figure 3).
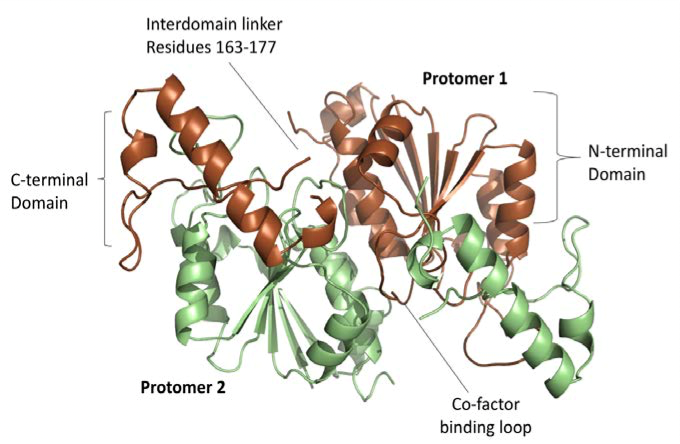
Figure 2: The 2 protomers of TrmD active site including the co-factor binding loop and the disordered interdomain linker.
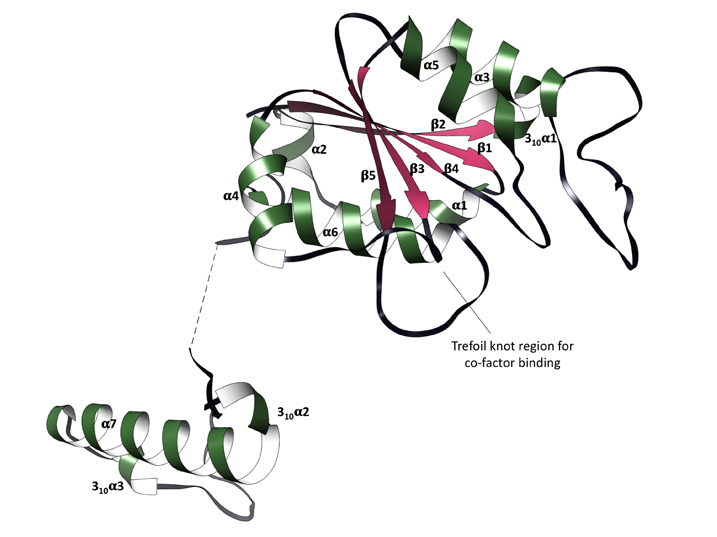
Figure 3: Trefoil knot motif (SAM and SAH binding site) containing 3 unfolded loops.
Through these crystal structures, several principal points were disclosed. Both SAM and SAH (un-methylated SAM) bind this trefoil knot in an L-shaped bent conformation and forms a network of hydrogen bonds, hydrophobic and pi-interactions. Primarily, the adenine ring in SAM and SAH forms hydrogen bonds with residues I133 and L138 in one loop of the trefoil knot, as well as G134 and Y136 in another loop (Figure 4).
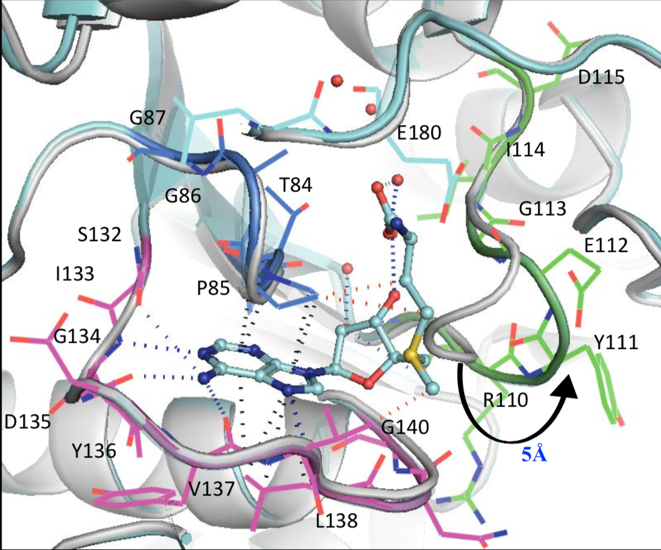
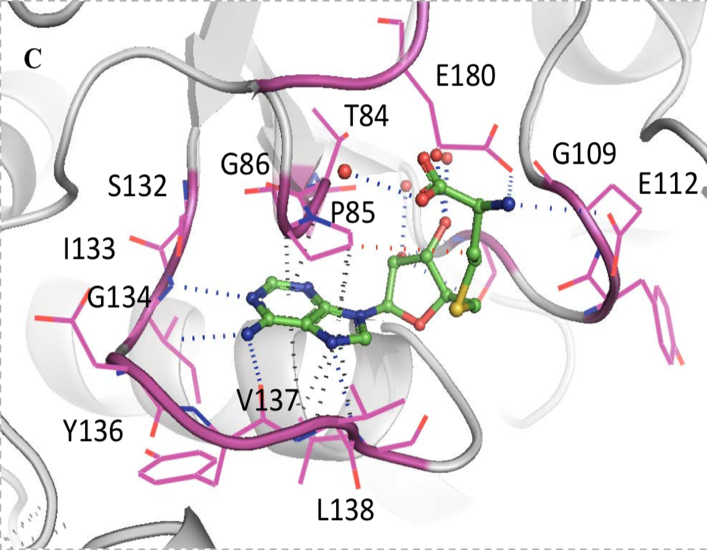
Figure 4: Network of intermolecular bonds between SAM and SAH with the loops of the trefoil knot motif of TrmD
Validated hits and viable fragments were divided into 3 clusters with distinct binding interactions
Based on the structural studies carried out on SAM and SAH binding in the active site of TrmD, 27 fragments resulted from a series of validation processes including the screening of 960 in-house library fragments and subsequent selection for X-ray crystallography experiments. The resulting fragments were categorized into 3 groups.
The first group encompasses fragments that only bind the region that holds the adenine ring of SAM in which various interactions (i.e. hydrogen bonds and pi-interactions) are formed with both aforementioned loops of the trefoil knot. The second group of fragments covers the ribose-binding pocket in addition to that of the adenine pocket. These fragments form interactions with all 3 loops. The third group involves fragments that also cover the methionine-binding site through additional hydrogen bonds.
Following categorization, fragments were merged for chemical optimisation, as seen in Figure 5, based on various criteria such as ligand efficiency, binding affinity and synthetic tractability.
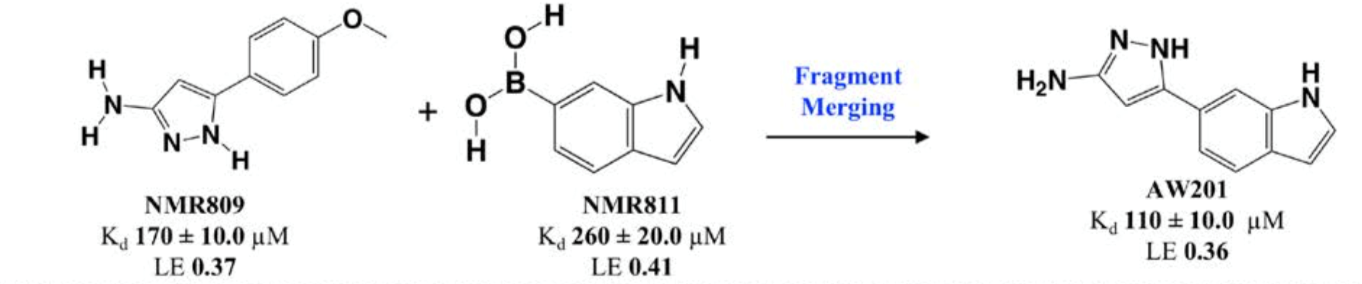
Figure 5: Reaction scheme of fragment merging.
2 of the 27 fragments were chosen for merging; one of these contains an aminopyrazole ring which forms a number of hydrogen bonds with T136, L138, G134 and S132, as well as hydrophobic interactions with the ribose binding site. The other fragment contains an indole ring which fits into the ribose pocket by forming a water-mediated interaction with L138 (Figure 6).
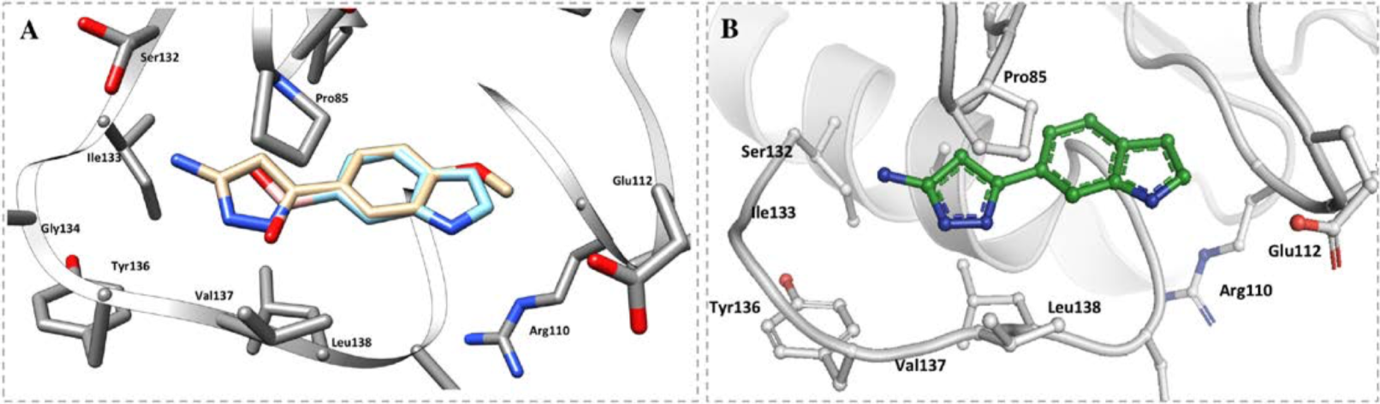
Figure 6: Binding model of A the 2 selected fragments, B) the compound made from the merged fragments in the adenosine pocket of TrmD
Lead compounds have anti-mycobacterial activity and kill intracellular M. abscessus and M. leprae
The effectiveness of the merged lead compounds on cytotoxicity for both healthy and M. abscessus-infected human macrophages was tested and compared.
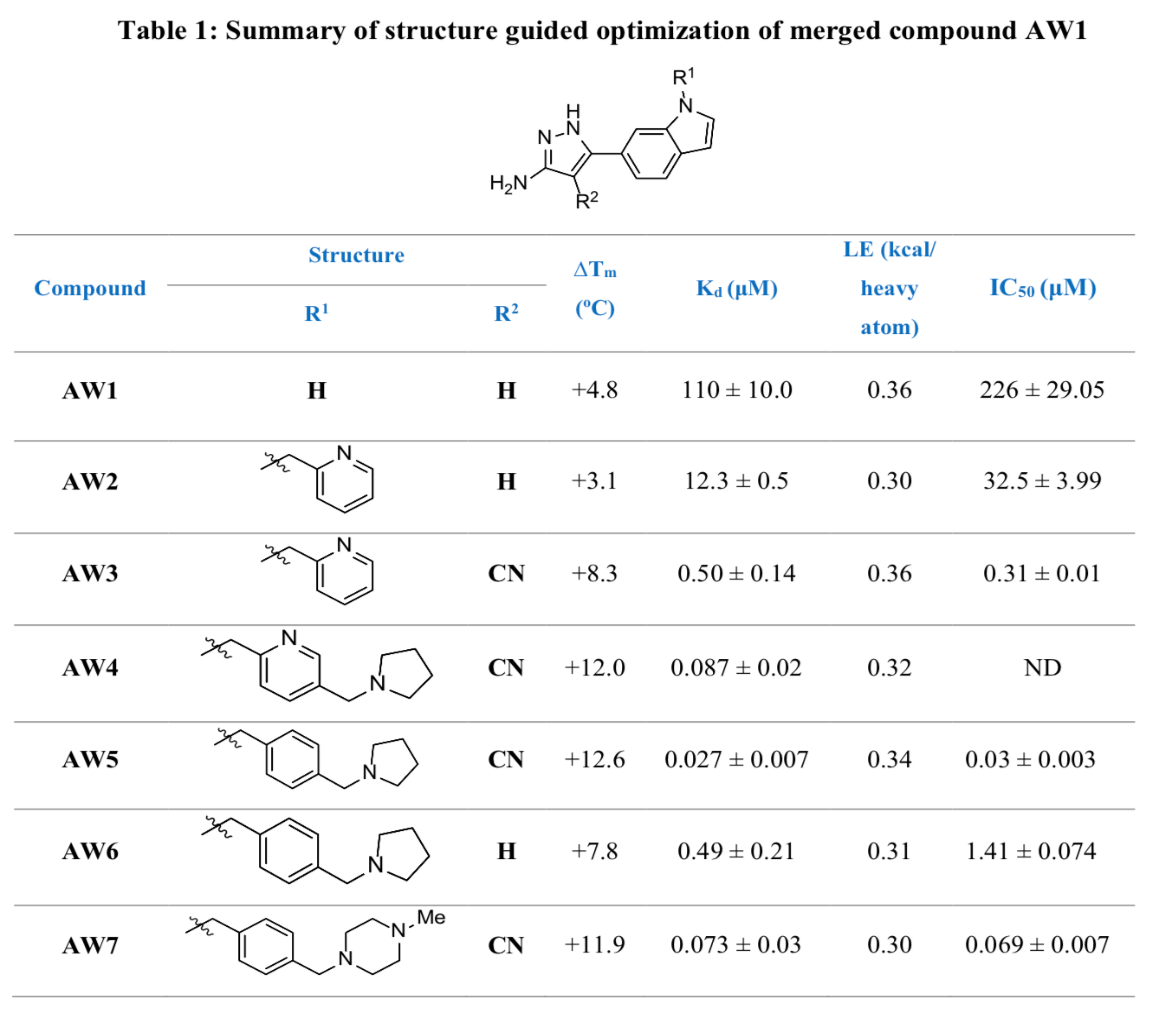
Both optimised lead compounds tested (AW6 and AW7 as seen in Table 1) demonstrated antibacterial activity, with AW7 leading to a higher reduction in colony-forming units than AW6 at the same concentration. Upon further testing, AW7 also demonstrated a much higher efficiency of inhibition to rifampicin against M. leprae at the same concentration.
Why I like this preprint
Coupled with the development of resistances against antibiotics, there is also a constant demand for the development of more effective drugs. With this in mind, there is a need to tackle the problems presented in drug resistance with novel and innovative approaches to achieve this. It is always interesting to explore alternative techniques used to optimise drug discovery in addition to traditional processes.
Questions for the authors
1. Apart from SAM/SAH, are there any other identified binding partners of TrmD or Trm5 that could be mimicked for drug development?
2. Compared to the traditional drug discovery process, in what ways is a fragment-based drug discovery approach more effective for drugs targeting active sites such as those in TrmD?
References
1. Bjork G.R., Wikstrom P.M., and Bystrom A.S. (1989). Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine, Science 244, 986-989.
2. Erlanson D.A., Fesik S.W., Hubbard R.E., Jahnke W., and Jhoti H. (2016). Twenty years on: the impact of fragments on drug discovery. Nature reviews Drug discovery 15, 605-619.
3. Mendes V. and Blundell T.L. (2016). Targeting tuberculosis using structure-guided fragment-based drug design. Drug discovery today.
doi: https://doi.org/10.1242/prelights.9189
Read preprint (No Ratings Yet)
(No Ratings Yet)