








HSP110 dependent HSP70 disaggregation machinery mediates prion-like propagation of amyloidogenic proteins in metazoa
Posted on: 27 October 2019 , updated on: 16 December 2019
Preprint posted on 9 October 2019
Article now published in The EMBO Journal at http://dx.doi.org/10.15252/embj.2019103954
Two sides of the same coin: Hsp110 activity protects against amorphous protein aggregation, but promotes amyloid aggregation and toxicity in C. elegans models
Selected by Tessa SinnigeIntroduction
Living cells and organisms depend on the function of molecular chaperones to ensure correct protein folding. In a variety of disease conditions and even during normal ageing, however, proteins can misfold and aggregate, posing a challenge to cellular protein homeostasis and leading to toxicity. For example, Alzheimer’s, Parkinson’s and Huntington’s diseases are all characterised by the deposition of protein aggregates in the brain, as are prion diseases. Yeast cells contain a particular chaperone, the Hsp104 disaggregase, which can solubilise existing aggregates. This activity may be beneficial, yet has also been shown to stimulate the propagation of yeast prions by generating smaller fibril fragments that can spread from cell to cell. Metazoa do not carry a Hsp104 homologue, but instead employ a complex of three chaperones, Hsp70/Hsp40/Hsp110, as a disaggregase that was shown to act on amorphous and amyloid-like aggregates in vitro (1, 2). In this preprint, the role of this disaggregase machinery in amyloid aggregation and the associated toxicity in vivo is investigated in the animal model C. elegans.
Key results
The authors of this preprint make use of well-established C. elegans models expressing the Parkinson’s disease related protein α-synuclein, and a fragment of the Huntingtin’s disease protein, polyglutamine, in the body wall muscle cells. Aggregation of the proteins in this tissue is associated with cellular toxicity that can be measured as a decline in the worms’ motility. The authors use an elegant system to achieve tissue-specific knock-down of Hsp110, which is part of the disaggregase machinery, to investigate the effects on protein aggregation and toxicity without causing widespread side effects.
First, they test the role of Hsp110 in amorphous aggregation using a luciferase reporter. The expression of luciferase in the worms leads to the formation of amorphous aggregates at higher temperatures, which disappear when the worms are returned to ambient temperatures. However, when Hsp110 is knocked down, a significant number of luciferase aggregates persists, confirming the role of Hsp110 in disaggregation. On the other hand, the knock-down of Hsp110 in the α-synuclein and polyglutamine worms results in a reduction in the number of foci and restores the motility defect. This result, although perhaps surprising at first sight, can be explained by the generation of smaller aggregate seeds catalysed by Hsp110, which overall leads to an amplification of aggregate formation by secondary processes. In support of this notion, the authors furthermore show that Hsp110 knock-down reduces the spread of α-synuclein to neighbouring tissue in a recently established C. elegans model (3), which is also thought to be mediated by small aggregate fragments.
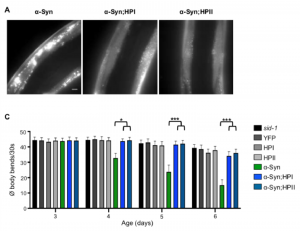
Although knocking down Hsp110 might sound like a good idea to halt disease-associated protein aggregation, this protein is critical to stimulate the ATPase activity of Hsp70, which in turn ensures the proper folding of a wide variety of client proteins. As such, a decrease in Hsp110 levels may interfere with protein folding and cellular homeostasis in general. Indeed, the authors find increased misfolding of a temperature-sensitive mutant muscle protein, accompanied by decreased motility, upon Hsp110 knock-down. Moreover, they find a faster decline in motility during ageing of wild-type animals when Hsp110 levels are lowered, consistent with an increased burden on the protein homeostasis capacity with age. In the case of the α-synuclein worms, this results in a loss of the protective effect of the Hsp110 knock-down beyond a certain age. Altogether, the ‘good’ and ‘bad’ activities of Hsp110 are thus two sides of the same coin, and the outcome will depend on the type of proteins that put the most pressure on the protein homeostasis system at a given time.
Why I chose this preprint
Disclaimer: I also work on protein aggregation using C. elegans models! I find this preprint a really great example of the beauty of this model organism to study the interplay between protein aggregation, toxicity and cellular quality control mechanisms. The findings are exciting because they shed more light on the function of Hsp110 and the metazoan disaggregase machinery. Furthermore, they show that amorphous and amyloid-like protein aggregation are two fundamentally distinct processes that need to be dealt with in different ways. Finally, the notion that chaperone activity can be ‘bad’, i.e. enhance the aggregation and spread of toxic proteins, is starting to gain more attention and will be very important when trying to target chaperones as a therapeutic strategy.
Questions
Can these results help us to understand the nature of the α-synuclein foci and the species that are capable of spreading? It has previously been shown that the majority of α-synuclein inclusions in one of these C. elegans models appear mobile and not amyloid-like (4, 5). Also the supposedly fibrillar nature of Lewy Bodies has recently come into question (6). Would the involvement of Hsp110 imply that α-synuclein inclusions must contain at least some amyloid fibrils, or could the disaggregase machinery also act on more disordered (intermediate) states?
Would an intervention to reduce Hsp110 activity be a promising therapeutic approach for amyloid-associated diseases? The effect on the general protein folding capacity may lead to negative consequences, but could this be a viable compromise in the context of a system challenged by amyloidogenic proteins?
References
- Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda NB, Szlachcic A, Guilbride DL, Saibil HR, Mayer MP and Bukau B (2015) Human Hsp70 Disaggregase Reverses Parkinson’s-Linked α-Synuclein Amyloid Fibrils. Mol. Cell 59: 781–793
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R, Guilbride DL, Wade RC, Morimoto RI, Mayer MP and Bukau B (2015) Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 524: 247–251
- Sandhof CA, Hoppe SO, Druffel-Augustin S, Gallrein C, Kirstein J, Voisine C and Nussbaum-Krammer C (2019) Reducing INS-IGF1 signaling protects against non-cell autonomous vesicle rupture caused by SNCA spreading. Autophagy 0: 1–22
- Van Ham TJ, Thijssen KL, Breitling R, Hofstra RMW, Plasterk RHA and Nollen EAA (2008) C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genet. 4: e1000027
- Laine RF, Sinnige T, Ma KY, Haack AJ, Poudel C, Gaida P, Curry N, Perni M, Nollen EAA, Dobson CM, Vendruscolo M, Kaminski Schierle GS and Kaminski CF (2019) Fast Fluorescence Lifetime Imaging Reveals the Aggregation Processes of α-Synuclein and Polyglutamine in Aging Caenorhabditis elegans. ACS Chem. Biol. 14: 1628–1636
- Shahmoradian SH, Genoud C, Graff-Meyer A, Hench J, Moors T, Schweighauser G, Wang J, Goldie KN, Suetterlin R, Castano-Diez D, Perez-Navarro P, Huisman E, Ipsen S, Ingrassia A, de Gier Y, Rozemuller AJM, Da Paepe A, Erny J, Staempfli A, et al. (2019) Lewy pathology in Parkinson’s disease consists of a crowded organellar membranous medley. bioRxiv 2019: 10.1101/137976
doi: https://doi.org/10.1242/prelights.14845
Read preprint (No Ratings Yet)
(No Ratings Yet)