








Modulation of neuronal resilience during aging by Hsp70/Hsp90/STI1 chaperone system
Posted on: 12 June 2019 , updated on: 17 June 2019
Preprint posted on 13 February 2019
The chaperone network as a highway to tissue resilience: the Hsp70/Hsp90 machinery prevents age-dependent hippocampal neurodegeneration
Selected by Giuliana ClementeIntroduction and background:
“You can chain me, you can torture me, you can even destroy this body, but you will never imprison my mind.”
In spite of the multiple times he got imprisoned, Gandhi endured with courage and determination a long peaceful protest to free India from foreign domination. Gandhi set an example of civil resilience, teaching how to patiently cope with crisis and how to find new and innovative ways to flourish again.
If one would like to draw a parallel between society and the human body, tissues are also continuously exposed to environmental insults which can lead to extensive damage to lipids, proteins and DNA. Whether or not a cell is resilient to stress-induced damage or goes down the route of an apoptotic response depends on the prompt activation of stress sensors and cytoprotective pathways.
Heat shock proteins (Hsp) Hsp70 and Hsp90 are two families of ubiquitously-expressed chaperones that, along with numerous co-chaperones, form an intricate network that assists protein folding and maturation (Figure1). This network plays therefore an important cytoprotective function within cells not only by taking care of the folding of newly forming polypeptides, but also by assisting the repair and refolding of damaged proteins and the degradation of irreparable ones.
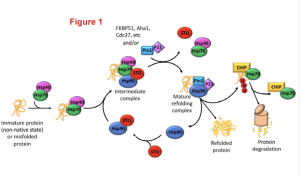
Figure 1: The Hsp70/Hsp90 machinery and the associated co-chaperones assist protein folding since from the early stage of protein synthesis. The figure highlights how the co-chaperone STl1(in red) bridges together Hsp70 and Hsp90 to smoothly transfer a client polypeptide from one complex to the other and to ensure its folding in a mature and functional protein. Figure was adopted from the preprint.
Hsp70 and Hsp90 work as a team and their activity is coordinated by a co-chaperone known as stress-inducible phosphoprotein 1 (STl1). STl1 is a modular protein containing tetratricopeptide domains (TPR1, TPR2A, TPR2B) which mediate the interaction between Hsp70 and Hsp90. Specifically, TPR1 and TPR2A bind to Hsp70 while TPR2B is responsible for the interaction with Hsp90, and thus acts as a molecular bridge. STl1 is required for the regulation of tissue homeostasis and resistance to stress in many model organisms (including mouse). However, the relevance of STl1 activity for brain function in general and especially during aging is still somewhat elusive, also due to the fact that STl1 knockout in mice is embryonic lethal. In this preprint, Lackie and colleagues engineered an hypomorphic STl1 allele to better investigate the functional requirement of STl1 in the chaperone network and evaluate its contribution to tissue resilience during aging.
A summary of the preprint:
To clarify the role of STl1 within the chaperone network in mammals, the authors generated a hypomorphic STl1 allele by Cre-lox recombination, removing exons 2 and 3 of the gene. The Cre-lox induced deletion removed the TPR1 domain (hereafter DTPR1 allele) and resulted in the expression of a smaller protein of 53kDa.
The DTPR1 STl1 mRNA was translated with a very poor efficiency and therefore STl1 protein levels were reduced by 80%. Most homozygous DTPR1 mice developed through embryogenesis, but 20% died after birth within the first month of life. This suggests that homozygous DTPR1 mice are mostly viable and that the truncated protein, even if expressed at very low levels, provides with enough function for survival.
The authors then evaluated whether reduction of STl1 in the brain tissue of DTPR1 model results in the changes to the Hsp70/Hsp90 network. Protein and mRNA levels of Hsp70 and Hsp90 as well as their subcellular localization were unaltered. Furthermore, the expression levels of the main regulators of the heat shock response did not significantly change, suggesting that the core unit of the Hsp70/Hsp90 network was unaltered. However, the authors found a 40, 50 and 75% reduction in the protein levels of CHIP (a Ubiquitin E3 ligase), Pin1 and CypA (peptidyl-prolyl cis/trans isomerases), respectively. This result suggests that STl1 is regulating the stability of important Hsp90 co-chaperones. The STl1-mediated regulation is probably occurring at the protein levels as no evident changes in the transcriptome profile of DTPR1 Stl1 mice were detected.
What is the functional consequence of expressing DTPR1 STl1 variant? The authors evaluated the expression levels of bona fine Hsp90 clients in cortex and hippocampal brain tissues from control and DTPR1 STl1 samples. They found that the levels of classical Hsp90 clients, including glucocorticoid receptor (GR), Tau protein and G protein coupled receptor kinase 2 (GRK2), were strongly reduced both in the cortex and in the hippocampus, suggesting that the activity of the Hsp70/Hsp90 network is likely to be crucial in the diverse regions of the brain.
As STl1 knockout is embryonic lethal it is not possible to formally test whether the effects observed in a DTPR1 STl1 background are similar to the ones that would be found in a STl1-KO background and to what extent. In an attempt to test this, the authors generated a STl1-KO cell line using CrispR-Cas9 technology and reported results comparable to the ones reported for the hypomorph allele, suggesting that the activity of the Hsp70/Hsp90 network strongly relies on Stl1 expression levels and activity. Moreover, they observed that cell proliferation and viability were significantly compromised upon STl1 knockout. Cells were in fact very sensitive to thapsigargin-induced ER stress, suggesting that they were generally less resistant to environmental stress. Picking up from this observation, they tested whether this result could be recapitulated in vivo. Neuron vulnerability to misfolding during aging was compared between young (3-5 months) and old (15 months) control and DTPR1 STl1 mice by counting the number of mature neurons in different brain subregions. A prominent thinning of CA1 and CA3 regions due to neuron loss was observed in brains of old animals compared to control. Interestingly, they also noticed that the levels of STl1 protein decreased significantly between old and young control mice. This means than in DTPR1 STl1 mice, where the levels of the proteins are already dramatically reduced compared to control, a further reduction in the expression levels predisposed animals to neurodegeneration. As a further proof, mice were challenged with the Morris Water Maze (MWM) test. Even though both control and DTPR1 STl1 mice were able to learn the MWM task, on the trail day the DTPR1 STl1 animals did not show any preference for the target quadrant, meaning they were unable to remember the information learnt. Therefore, neuronal degeneration in the CA3 region was accompanied in by a selective loss of spatial memory.
Why is this preprint relevant?
This study shows how powerful can be the generation of a hypomorphic allele can be to describe the functional relevance of a protein during vertebrate development. Specifically, the work identifies in STl1 the master regulator of the Hs70/Hsp90 network which controls the stability of some Hsp90 client proteins and important co-chaperones. Moreover, this study underlines how important Stl1 is for ensuring the physiological aging of the brain as optimal activity of STl1 is critical for neurons to effectively deal with increased stress during aging and for the proper removal of senescent cells.
Questions to the authors:
- As STl1 can be secreted, has the activity of the DTPR1 STl1 variant been tested in the extracellular space? Is there an increased accumulation of abaggregates?
- Would conditional overexpression of STl1 in wildtype control mice delay aging or you would expect it to be equally deleterious?
- As heterozygous mice show similar reduction in protein levels, do they also show similar neurodegeneration with aging? Do they perform as the homozygous mutants at the behavioral tests?
- The data presented in this work suggest that STl1 could be potentially used to selectively regulate the activity of Hsp90 helping cells to finetune the activity of this important chaperone. Is there any data on the number of SNPs at this location and whether any of these correlate to the increase likelihood of developing neurodegenerative diseases and/or other disorders?
doi: https://doi.org/10.1242/prelights.11172
Read preprint (No Ratings Yet)
(No Ratings Yet)