








Molecular recording of mammalian embryogenesis
Posted on: 23 August 2018
Preprint posted on 3 August 2018
Article now published in Nature at http://dx.doi.org/10.1038/s41586-019-1184-5
Background
Understanding how pluripotent cells give rise to the myriad of cell types required for building a complex adult organism is a central goal in developmental biology. Recent advances in single-cell RNA-seq (scRNA-seq) technologies has meant that the fates of individual pluripotent cells can now be followed at the transcriptional level to understand the embryonic origins of adult tissues. Recently, some very exciting papers were published together in the journal Science, whereby fate maps for frog, fish and planarian were generated by sequencing tens of thousands of individual cells at different stages of development [1-4]. To follow the fates of individual cells over time, unique heritable synthetic DNA ‘barcodes’ were incorporated into early embryos, and used in conjunction with scRNA-seq data to describe the origins of different cell types and the relationships between them.
So far, generating fate maps for mammalian development has been harder, because of technical difficulties in collecting a range of exact age-matched embryos, and because of the much longer duration of their development relative to fish and frog. However, earlier this month the Weismann lab posted a preprint describing and validating their methods for constructing an incredibly detailed fate map of early mouse development, using CRISPR-Cas9 to make continually evolving lineage tracer.
How it works
As with all CRISPR-Cas9 based technologies, the author’s toolkit comprises guide RNAs and Cas9, which cut at their target sequence – in this case the lineage tracer, or target site cassette shown below (Figure 1):
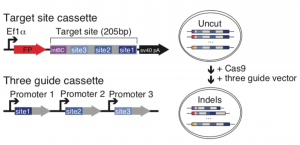
Figure 1. Schematic diagram of target site construction and guide cassette (A) and indel formation (B) taken from Figure 1 of the preprint, reproduced under CC-BY-NC-ND 4.0.
The target site cassette is designed with an 8bp static integration barcode (intBC), followed by three different CRISPR target cut sites. Adapting an approach already used for lineage tracing in zebrafish [5-6], these CRISPR target sites are engineered to have variable Cas9 cutting rates. After Cas9 induces a DNA strand break in the target site cassette, insertions and deletions (indels) are introduced during the repair process, with some sites accumulating indels over a period of hours, and others over a span of days to weeks. Therefore, cells acquire a unique indel or barcode signature which irreversibly changes over time, enabling molecular recording of individual cells throughout development. During single-cell sequencing, the transcriptome of each cell is sequenced alongside the unique indel signature, which serves as a readout for lineage tracing – cells containing a particular intBC must share the same pluripotent ancestor, and those containing more similar indel signatures being more likely to be part of the same cell lineage.
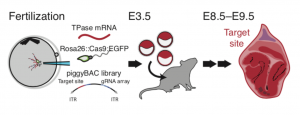
Figure 2. Schematic diagram of lineage tracing in mouse, taken from Figure 2 of the preprint, reproduced under CC-BY-NC-ND 4.0.
To apply this in vivo, the three guide cassette vector and a library of target site cassettes were incorporated into mouse oocytes by PiggyBac retrotransposition, as shown in Figure 2. The oocytes were fertilised by sperm carrying a form of Cas9:GFP, which is activated at the onset of zygotic transcription. The embryos were cultured in vitro for 96 hours before being transferred to a surrogate female mouse for a week to develop to stage E8.5. They were then processed for scRNA-seq.
Key findings
The authors first performed several preliminary experiments to show that their lineage tracer could follow multiple cell populations simultaneously, and that indel signatures varied sufficiently between bulk tissue samples of placenta and embryonic regions, which should have diverged early in development.
They then performed scRNA-seq and confirmed their lineage tracing system was fit for purpose, as up to 75% of cells recovered containing intBCs, and the lineage tracer generated enough sequence diversity throughout the experiment. Next, the authors analysed their scRNA-seq data more conventionally to cluster cells based on their transcriptional profile, thereby assigning them a function and temporal identity. By overlapping these clusters with their lineage tracer indel signatures, their relationships to each other could be observed. For example, placental and visceral endoderm cells were most distantly related to other clusters, and neural plate and ectoderm tissues more closely related, which recapitulates known relationships between developing tissues.
The authors then constructed a fate map of mammalian development at the single cell resolution, by using the changing sequence of the lineage tracer to reconstruct phylogenetic cell lineage trees. All cells captured at E8.5 represent the tips of a tree branch, and all cells sharing a common intBC signature must have derived from the same founder ‘root’. By comparing first more closely related, and then more distantly related indel signatures, branch points between different cell lineages could be inferred further and further back in time. In this way, all cells could be assigned positions in different cell lineage trees. From this data, the authors could infer that 1-6 totipotent cells, 4-17 early and 15-52 late pluripotent progenitors gave rise to all cells collected at E8.5 – a surprising variability suggesting many backup options are required to assemble a functional organism.
To test the fidelity of the lineage trees, the authors mapped the transcriptional scRNA-seq information onto each reconstructed lineage. Generally, the reconstructed tissue relationships correlated well with known knowledge of development, and cells with highly similar indel signatures had more similar transcriptional profiles. From this information, the authors also found unexpected tissue relationships. For example, a population of lineage-traced cells assigned to the embryonic endoderm had a transcriptional identity characteristic of extraembryonic endoderm, expected to have diverged much earlier in development. This suggests that maybe independent cell lineages can converge, and that this lineage tracing approach can potentially uncover lineage plasticity and heterogeneity that could be missed using transcriptional analyses alone.
Why I chose this preprint
Given how complex and diverse animals are, I find it fascinating how we all started life as a ball of fairly uniform stem cells, and admire ambitious studies like these which aim to understand developmental processes on a global level. As single-cell sequencing is such a new technology, and with the inherent complexity of studying mammalian development, I thought that it would take a longer time before mammalian fate mapping caught up with other vertebrates. Therefore, I think this work is a big achievement in providing a toolkit to study this, and other similarly complicated processes. Even though at this stage in the data analysis, this work does not tell us much that we did not already know about mammalian development, it starts to suggest that developmental processes may not be as linear and predetermined as previously thought, which might change how we think about cell signalling and organisation during development.
Future directions
Although used in this case for studying a specific timeframe of development, the techniques outlined in this preprint can be employed for many situations. By varying the lineage tracer target site sequences, the mutation rate can be tuned to follow more long-term processes, or by altering the Cas9, lineage recording in specific compartments should be possible in development and regeneration, or in response to stress or action potentials. It would also be exciting to see if this could be used in cancer studies to study how subpopulations of transplanted tumour cells behave over time.
References
- Fincher, C.T. et al., 2018. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science, 360(6391)
- Farrel, J.A. et al., 2018. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science. 360(6392)
- Briggs, J.A. et al, 2018. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. 360(6392)
- Wagner, D.E et al 2018. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. 360(6392)
- McKenna, A. et al., 2016. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science, 353(6298)
- Spanjaard, B. et al., 2018. Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nature Biotechnology. 36(5)
doi: https://doi.org/10.1242/prelights.4451
Read preprint (No Ratings Yet)
(No Ratings Yet)