








Systematic identification of human SNPs affecting regulatory element activity
Posted on: 29 November 2018 , updated on: 30 November 2018
Preprint posted on 4 November 2018
Article now published in Nature Genetics at http://dx.doi.org/10.1038/s41588-019-0455-2
Assessing the regulatory potential of human genetic variation: use of Massively Parallel Reporter Assays to identify >30,000 SNPs that modulate reporter expression.
Selected by Jesus VictorinoBackground & Summary:
Since Genome-Wide Association Studies (GWAS) began to highlight genetic risk loci linked to common diseases almost a dozen years ago (1), much effort has been devoted to elucidating the functional role of these Single Nucleotide Polymorphims (SNPs). Strinkingly (at that time), most of these common genetic variants in the human genome were non-coding, which led to the study of their potential contribution to gene regulation. In recent years, Massively Parallel Reporter Assays (MPRAs) have allowed to assess promoter or enhancer activity in thousands of DNA fragments en masse moving the genomics field forward (2).
In this preprint, the authors interrogate the genome almost at its entirety for its ability to control gene expression by means of MPRAs. Thanks to a >100-fold scale-up, they are able to test millions of randomly fragmented DNA sequences coming from 4 divergent genomes which results in libraries that contain both alleles for nearly 6 million SNPs. After they performed this ‘even-higher’-throughput assay in two different cell lines, Joris van Arensbergen and colleagues identified >30,000 SNPs that altered enhancer activity mostly in a cell-type specific manner.
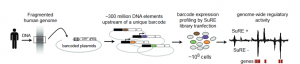
Fig.1. Experimental design used by van Arensbergen et al. to identify enhancer elements (Image taken from figure 1a).
Why I chose this work:
The readout of current genomic approaches such as DNase-seq, ATAC-seq or ChIP-seq for some histone modifications are widely used as surrogate markers for active regulatory elements, that control gene expression in a tissue-specific manner. This is due to the fact that, to date, MPRAs do not produce genome-wide maps and, therefore, sometimes open chromatin and histone mark signatures are the best deals one can get. Hypotheses based on such predictive models need to be, in any case, further confirmed.
I chose this preprint for its three major contributions to help understanding the non-coding genome:
- The authors overcame MPRA size limitations to study mammalian large genomes and they were able to provide a whole-genome snapshot of enhancer activity.
- Thanks to the use of four divergent genomes, they could test both major and minor alleles for >50% of all common SNPs in a hepatocarcinoma and a myeloid leukemia cell line. This is extremely useful to understand the effect of human variation both in homeostasis and diseased state such as hepatic or blood cell disorders.
- They also integrated enhancer activity data with eQTLs and GWAS information helping prioritization of candidate causal genes. As an example, the authors found that variant rs3788853 stands out by showing a ~5-fold effect among 30 eQTLs associated to the angliodema-related XPNPEP2 gene expression.
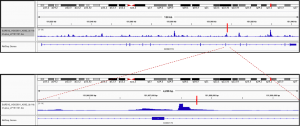
Fig2. Enhancer activity map of a genomic region snapshot containing the Chronic Myeloid Leukemia (CML)-associated variant rs4869742 in K562 cells. SNP is marked by a vertical red bar. Obtained from the available online data in OSF (https://osf.io/w5bzq/).
At the same time, tissue-specificity of enhancers is thought to be encoded at the sequence level but made effective by the binding of transcription factors (TFs). However, TF ChIP-seq data have shown that affinity for TFBS, although sequence-dependent, does not necessarily need to correlate with functionality. The authors analyzed the disruption of predicted TFBS in alleles that showed an impact on enhancer activity providing a list of putative functional sites for TFs. At rs623853, they experimentally found a loss of binding of Ets-like factors specifically in one allele, contributing to the causal relationship between eQTLs and GWAS SNPs.
Scientific relevance for the field:
The non-coding genome is a fascinating >98% of our genetic material that needs to contain the detailed information to control gene expression precisely in time and space. Despite the rise of ‘omics approaches and the flood of big data that come with it, yet very little is known about the enhancer repertoire in the human genome. Since the technology did not allow it, there was nothing like a whole-genome enhancer map in vertebrates. MPRAs have partially filled this gap by functionally assaying prioritized loci or variants and have nicely contributed to narrowing down causal SNPs in disease-relevant contexts. Nonetheless, the use of different technology and approaches in different labs makes the unification and display of the data in a single version tricky.
With this work, van Arensbergen et al. have generated genome-wide maps of enhancer activity in a single assay for two different cell lines. Having these datasets available will be a useful resource for any scientist studying gene regulation or how human genetic variation contributes to its control, even with MPRAs’ existing limitations due to their plasmid-based technology in which genomic regions are tested ‘out of context’.
Questions to the authors:
- Having genome-wide maps of enhancer activity will be extremely useful but, what do the authors think about the resolution at which one can look at a particular locus? Will it still be better having less complex libraries for that?
- SuRE is defined as a promoter-less system that can be used to test enhancers and promoters. Are SuRE values for promoters higher than those for enhancers? Do the authors think that promoter activity detected in the assay might mask the detection of enhancers?
- In this preprint the authors claimed that a feature of SuRE is that it outperforms current MPRA technology in terms of library complexity by >100-fold. Do the authors think that other unbiased systems that use fragmented DNA (e.g. STARR-seq) are not able to reach the same degree of complexity?
- Since previous MPRA studies have measured enhancer activity in human cells, how well does SuRE compare to other existing MPRA systems at equivalent loci?
- The genomics field focuses on identifying active elements by direct methods such as MPRAs, however the identification of repressing elements is mainly predicted through indirect measurements such as the presence of some histone marks and/or members of repressing complexes. How do the authors envision the direct detection of repressor elements systematically?
Reference:
- Wellcome Trust Case Control Consortium. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447, 661-678.
- Inoue F., Ahituv N. 2015. Decoding enhancers using massively parallel reporter assays. Genomics. 3, 159-164.
doi: https://doi.org/10.1242/prelights.6046
Read preprint (3 votes)
(3 votes)