








Alteration of cardiolipin-dependent mitochondrial coupling in muscle protects against obesity
Posted on: 4 November 2019 , updated on: 8 November 2019
Preprint posted on 26 July 2019
A mitochondrial defect with unexpected consequences. Reduced cardiolipin content in mitochondrial membranes impairs phosphorylating respiration but protects Hacd1-deficient mice against diet-induced obesity
Selected by Pablo Ranea RoblesBackground
Mitochondrial function is particularly important in skeletal muscles, which consume a lot of energy for their contraction. The curved structure of mitochondrial cristae is modelled by the lipid composition of the inner mitochondrial membrane (IMM). Cardiolipin is a phospholipid with four acyl chains in its structure, that exerts a major role in the curvature of the IMM. It provides stability and fluidity to the IMM, and its interaction with several components of the mitochondrial electron transport chain is critical for its optimal function (Paradies et al., 2014). Remodeling of cardiolipin with C18 fatty acids by Tafazzin is essential for its normal function. This is exemplified by Barth syndrome, which is caused by mutations in Tafazzin (Barth et al., 1983). Fatty acids with 18 carbons or more are generated by the very long chain fatty acid elongation cycle, which consists of four reactions of condensation, reduction, dehydration, and reduction. HACD1 catalyzes the third step, in which it dehydrates 3-hydroxyacyl-CoA, generating 2,3-trans-enoyl-CoA. Mice and dogs deficient in HACD1 (3-hydroxyacyl-CoA Dehydratase 1) show reduced muscle mass and strength (Blondelle et al., 2015; Pelé et al., 2005). However, how HACD1 deficiency impacts muscle function and metabolism remain still unknown.
Main findings
In this preprint, Prola and colleagues (Prola et al., 2019) report that HACD1 deficiency leads to a reduction in cardiolipin content in skeletal muscle. This deficit in cardiolipin caused dilation of mitochondrial cristae and impaired phosphorylating respiration. Surprisingly, when fed with a high-fat diet (HFD), energy expenditure in HACD1-deficient mice was increased, protecting them against the deleterious effects of obesity (Fig. 1).
The key to the muscle phenotype in Hacd1-deficient mice and dogs resides in changes to their mitochondria. When observed under the electron microscope, some of the Hacd1 KO mitochondria presented thickened cristae. However, the authors did not find any alterations in ATP synthase dimers or MICOS oligomers, which are involved in the maintenance of the cristae structure. They did not observe any change in the assembly of supercomplexes either, although the data is not shown in the preprint, and would be interesting to see. Altogether, these data pointed to a change in the lipid composition in the IMM.
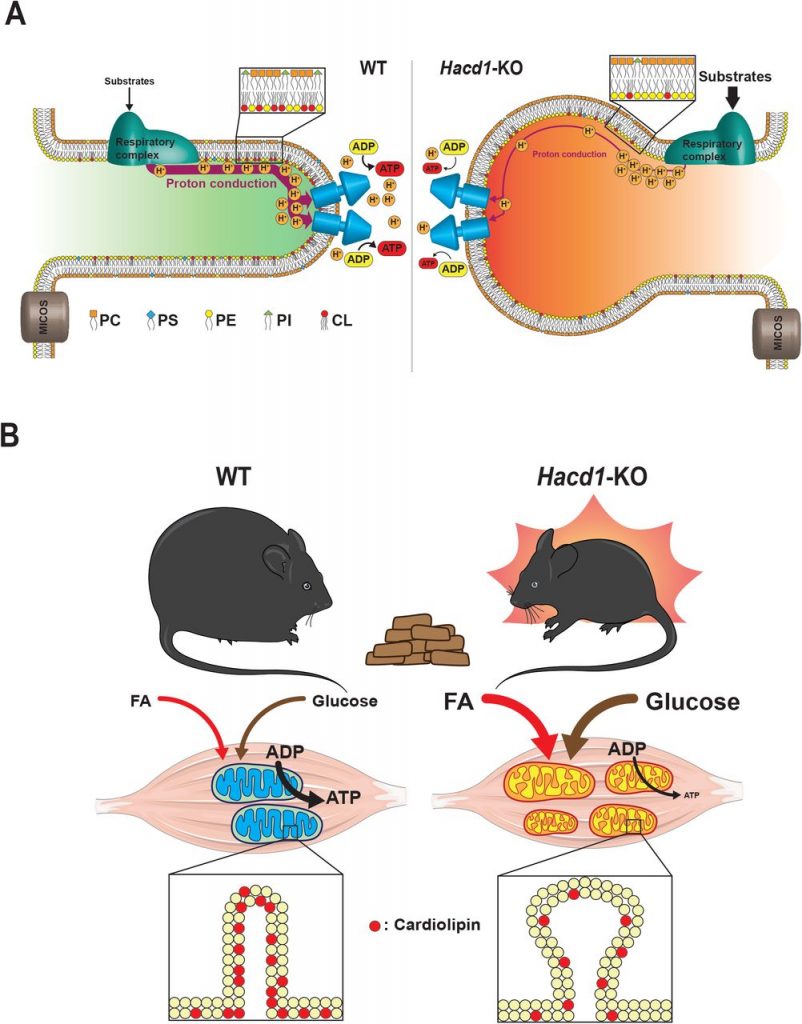
Figure 1. Graphical summary of Prola et al. study. A) Hacd1 deficiency led to a reduction in cardiolipin content in mitochondrial membranes that impaired mitochondrial phosphorylating respiration. B) Hacd1 KO mice were protected against diet-induced obesity due to increased energy expenditure despite an impaired mitochondrial phosphorylating respiration. Taken from Figure 7 of the preprint, made available under a CC-BY-NC-ND 4.0 International license.
HACD1 deficiency in mice caused a reduction in phosphorylating respiration in isolated mitochondria. There are several possible explanations for this result, but the authors discarded protein uncoupling or impaired function of the respiratory chain. Thus, this defect is likely due to an intrinsic membrane defect. This is shown by a difference in TMA-DPH fluorescence lifetime decay between WT and Hacd1-KO mice. TMA-DPH is a fluorescent probe used for measuring membrane fluidity in artificial and living membrane systems. They found that the most prominent difference was in the longest lifetime components, which corresponds to the most polar domains. This result was accompanied by a reduction in cardiolipin and total phospholipid content in the skeletal muscles of Hacd1-KO mice. Moreover, there was a reduction in the C18-C26 to C10-C17 ratio, suggesting that fatty acid elongation is impaired in Hacd1-KO mice. Again, the data suggest that a change in the lipid composition of the IMM, in particular, a reduction in the cardiolipin content, is responsible for the muscle defect. To prove the role of cardiolipin, they enriched muscle mitochondria with cardiolipin in the form of small unilamellar vesicles that fuse with mitochondrial membranes. O2 consumption and ATP production decreased in WT and Hacd1 KO mitochondria when cardiolipin was added, but the phosphorylating respiration deficit was fully rescued.
Hacd1 KO mice displayed a remarkable phenotype, considering the reduced cardiolipin content and the mitochondrial abnormalities observed in their muscles. Their endurance capacity was reduced due to the muscle hypotrophy, but glucose handling was surprisingly better compared to WT mice. They were more tolerant to glucose and had a higher insulin sensitivity. Even though they were less active than their WT littermates, energy expenditure was increased in Hacd1 KO mice. The authors then stressed WT and Hacd1 KO mice with HFD. Again, energy expenditure was higher in Hacd1 KO mice, which pointed to a better adaptation to HFD. This would suggest enhanced fatty acid oxidation in Hacd1 KO mice fed with HFD. Respiratory exchange ratio (RER) was decreased during the night in Hacd1 KO mice fed with a normal diet. RER is the ratio of CO2 production to O2 consumption and indicates the primary metabolic fuel (carbohydrates or fatty acids). During the night, fatty acids are not the primary metabolic fuel to produce energy, so the reduced RER suggest that fatty acids are the preferred metabolic source in Hacd1 KO mice. Moreover, the authors found that mitochondrial biogenesis was increased in Hacd1 KO mice. Altogether, they point to a shift to oxidative metabolism in the muscles of Hacd1 KO despite a decrease in mitochondrial coupling efficiency.
Probably the most striking result of this work is the protection against diet-induced obesity observed in Hacd1 KO mice. This protection was achieved even though Hacd1 KO animals ate more food than their WT littermates. Hacd1 KO mice fed with HFD retained their higher glucose tolerance and sensitivity to insulin and were resistant to muscle and liver steatosis. The most plausible explanation for this protection is the increased energy expenditure observed in the Hacd1 KO mice.
In summary, Prola and colleagues describe a novel role of cardiolipin in mitochondrial coupling. They discovered this role in Hacd1 KO mice, which had less cardiolipin in mitochondrial membranes in the muscle. This reduction in cardiolipin content caused mitochondrial abnormalities and a reduction in mitochondrial coupling efficiency. However, this muscle-specific cardiolipin deficit was associated with an elevation in basal energy expenditure and increased carbohydrate and lipid catabolism. This metabolic change protected Hacd1 KO mice against diet-induced obesity. These results suggest that targeting mitochondrial coupling efficiency could be explored as a therapy against metabolic disorders.
Why I picked this preprint:
I’m interested in the role of fatty acid metabolizing organelles such as mitochondria and peroxisomes in metabolic diseases. This preprint challenges the classical view in which a reduction in mitochondrial coupling is detrimental for metabolic health. A skeletal muscle-specific defect in mitochondrial coupling due to a deficit of cardiolipin in mitochondrial membranes increased basal energy expenditure. This happened despite the locomotor disability that Hacd1 KO mice suffered due to a loss in muscle mass. Another strong point of this work is that they developed a method to alter the composition of mitochondrial membranes that could be used by many labs in the field. Overall, the results obtained by Prola and colleagues are of interest to the metabolism field and deserve further discussion.
Questions for the authors:
- The response against HFD is remarkable in Hacd1 KO animals, but there is no difference in FAO between WT and Hacd1 KO mice, neither during daylight nor during the night period. Should not this be expected if they respond better against the high content of fat present in the HFD diet?
- You found an increase in mitochondrial mass by measuring different mitochondrial protein content, but is there any difference in mtDNA levels in the muscle of Hacd1 KO mice versus WT mice?
- Cardiolipin is particularly prone to oxidative attack. Do you discard cardiolipin oxidation as a cause for its depletion? Is there increased apoptosis in the Hacd1 KO muscle?
- Have you thought in any way of decreasing mitochondrial coupling that could be administered in vivo to test it in obesity or metabolic syndrome?
References
Barth, P. G., Scholte, H. R., Berden, J. A., Van der Klei-Van Moorsel, J. M., Luyt-Houwen, I. E., Van ’t Veer-Korthof, E. T., Van der Harten, J. J. and Sobotka-Plojhar, M. A. (1983). An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355.
Blondelle, J., Ohno, Y., Gache, V., Guyot, S., Storck, S., Blanchard-Gutton, N., Barthélémy, I., Walmsley, G., Rahier, A., Gadin, S., et al. (2015). HACD1, a regulator of membrane composition and fluidity, promotes myoblast fusion and skeletal muscle growth. J. Mol. Cell Biol. 7, 429–440.
Paradies, G., Paradies, V., De Benedictis, V., Ruggiero, F. M. and Petrosillo, G. (2014). Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta BBA – Bioenerg. 1837, 408–417.
Pelé, M., Tiret, L., Kessler, J.-L., Blot, S. and Panthier, J.-J. (2005). SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum. Mol. Genet. 14, 1417–1427.
Prola, A., Blondelle, J., Vandestienne, A., Piquereau, J., Denis, R. G., Guyot, S., Chauvin, H., Mourier, A., Letheule, M., Maurer, M., et al. (2019). Alteration of cardiolipin-dependent mitochondrial coupling in muscle protects against obesity. bioRxiv 715953.
doi: https://doi.org/10.1242/prelights.14944
Read preprint (2 votes)
(2 votes)