








Dissection of the Fgf8 regulatory landscape by in vivo CRISPR-editing reveals extensive inter- and intra-enhancer redundancy
Posted on: 25 March 2020
Preprint posted on 3 March 2020
Article now published in Nature Communications at http://dx.doi.org/10.1038/s41467-020-20714-y
Background
The development of an organism requires precise spatiotemporal regulation of gene expression, which is thought to be coordinated by enhancers. However, enhancers have been difficult to identify and study because they can be located up to megabases away from their target genes. Enhancer function is traditionally defined by its activity in plasmid reporter assays, which are often not representative of their endogenous function. Furthermore, there are often multiple enhancers for any given gene. Thus, it is crucial to understand the contribution of each individual enhancer to gene expression and to learn how they work together in vivo during development. In this preprint, the authors systematically make mutations to regulatory regions in the Fgf8 locus in mice to understand the contribution of each individual enhancer and to understand how the enhancers function together.
Key findings
FGF8 is a signaling molecule that is essential for the normal development of multiple tissues including the limb apical ectodermal ridge (AER) and the midbrain-hindbrain boundary (MHB). Putative enhancers regulating FGF8 expression in these regions have been identified in a previous study (Figure 1). To study the contribution of each enhancer, the authors used CRISPR/Cas9 to generate mice with hemizygous deletions of individual enhancers or combination of enhancers over Fgf8 null alleles.
For the limb AER enhancers, previous experiments have shown the deletions of enhancers CE58, CE59, CE61 and CE66 (Figure 1) leads to loss of limb Fgf8 expression and limb defects that mirror Fgf8 knockout mice. However, deletion of each individual enhancer leads to no changes in expression levels or pattern of Fgf8 and no visible phenotypic defect during development. The observation that each enhancer itself is dispensable but the collection of enhancers is important strongly suggests that these enhancers are functionally redundant.
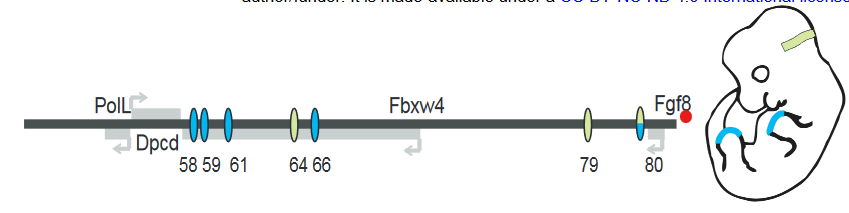
Figure 1: Schematic of Fgf8 regulatory locus. Blue represent limb AER enhancers and green represents MHB enhancers. Adapted from Figure 1A.
In the MHB, deletion of CE79 (DEL79), CE80 (DEL80) or both (DEL79-80) also appear to have no morphological defect or changes to the Fgf8 expression pattern. However, small but significant decreases in Fgf8 expression was detected in DEL80 and DEL79-80 animals. In addition, a panel of other genes in the MHB regulatory network showed a similar decrease, suggesting that there is a minor disruption to the MHB network.
In contrast, deletion of CE64 (DEL64) leads to a complete absence of midbrain and cerebellar structures at e8.25 and e18.5, phenocopying Fgf8 knockout mice. Thus, only one enhancer appears to be essential for the initiation and maintenance of Fgf8 expression in the MHB and therefore proper MHB development.
The authors then sought to dissect the regulatory logic of the CE64 enhancer using a CRISPR/Cas9 screen in mice embryos. They designed a set of CRISPR gRNAs targeting different parts of the CE64 enhancer and injected different pairs into oocytes that had been fertilized by males heterozygous for the DEL(P-58) allele (a non-functioning Fgf8 allele) to sensitise the screening background. Since CE64 disruption leads to severe phenotypic defect, the authors directly screened F0 embryos at e18.5 for loss of midbrain and cerebellum tissue. This allowed the authors to generate a relatively large set of overlapping deletions in vivo at the endogenous locus (Figure 2A). They identified three distinct elements (64-A, 64-B and 64-C) at the CE64 enhancer (Figure 2A and B). 64-A appears to be dispensable, while 64-B and C are essential for midbrain and anterior hindbrain development. However, deletions of smaller regions within 64-B were insufficient to disrupt function, suggesting that there is extensive functional redundancy in 64-B. In 64-C, deletion of a 37bp region was sufficient to completely render CE64 non-functional but deleting parts of this 37bp region does not have any effect, indicating that the 37bp contains at least two critical but redundant elements. However, neither 64-B or 64-C is sufficient to drive activity in the MHB in transgenic reporter assays.
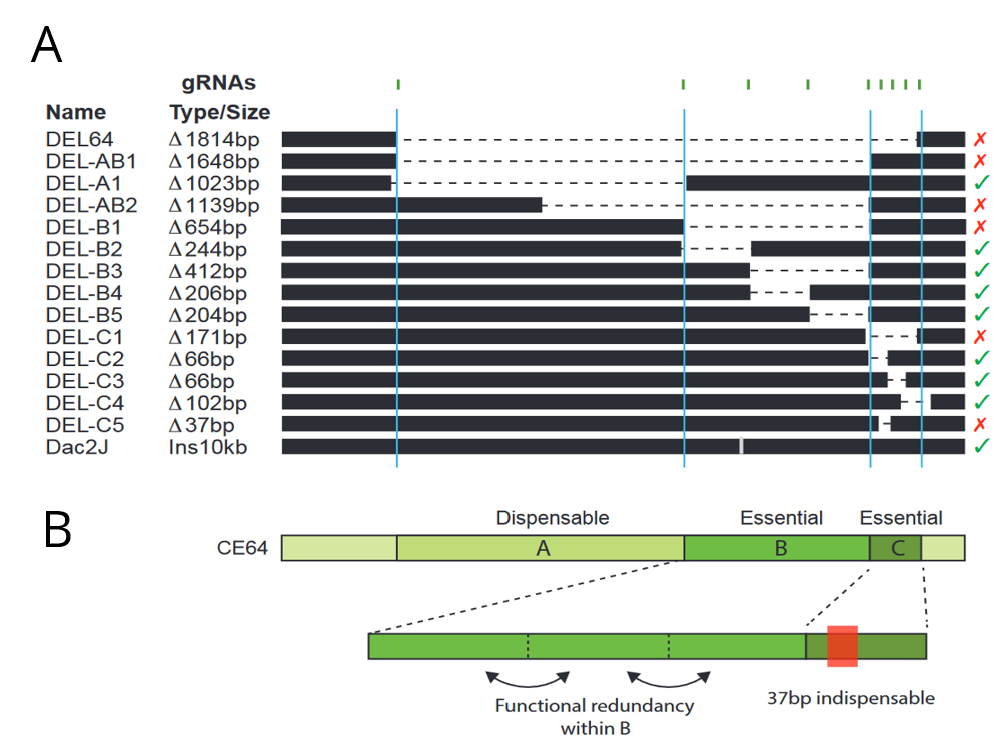
Figure 2: (A) Schematic of deletions made at the CE64 locus. The crosses and checks on the right indicate if function was conserved with the deletion. (B) Inferred structure of the CE64 regulatory locus. Adapted from Figures 4d and e.
The authors next examined how evolutionary conservation relates to function. It has been previously shown that the CE64 ortholog in zebrafish can drive expression in the zebrafish MHB, suggesting that CE64 function is conserved between mice and fish. While the 64-B region is highly conserved in fish, the 64-C region is not, even though 64-C was shown to be essential in mice. However, the CE64 region from spotted gar (a fish species closer to mouse) was sufficient to drive expression in the MHB in mice, though it did not completely reproduce its full expression. Because the 64-B region was not sufficient to drive transgenic gene expression, this suggests that non-conserved enhancer regions in fish can actually recapitulate similar expression patterns in mice.
Finally, the authors identified 4 conserved blocks of sequence in 64-C and found putative transcription factor binding sites (TFBS) within those blocks (Figure 3). Two of these blocks are disrupted by the deletion of the essential 37bp in 64-C, suggesting that these transcription factors are crucial for regulation. Furthermore, the block #3 position weight matrix (PWM) was found in spotted gar, while the block #2 PWM was found in zebrafish. These results suggest that while the complete sequences are not conserved, functional units might still be conserved in different positions.
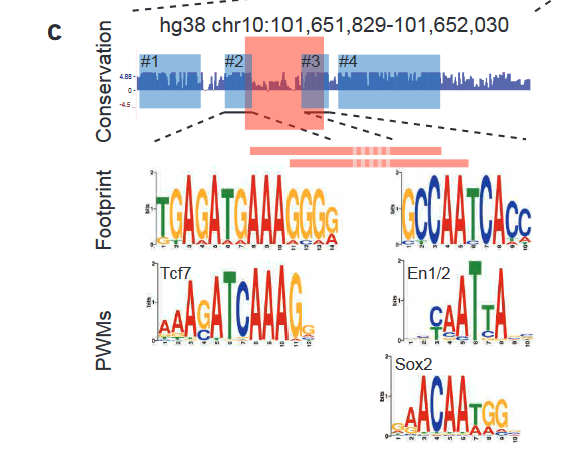
Figure 3: 4 conserved blocks of sequence in 64-C region and the corresponding putative TFBS. Adapted from Figure 6C.
What I liked
This is a massive study of the Fgf8 regulatory logic in vivo at the endogenous Fgf8 locus, which is pretty impressive in itself. The genetic ‘enhancer bashing’ in vivo is, in my opinion, a really nice combination of old school genetics and new techniques. It has been unclear how much redundancy exists between enhancers and how multiple enhancers act together to regulate a gene. The results seem to suggest that even for the same gene, different tissues can use different logic to regulate expression – for example, highly redundant enhancers in the limb and one essential enhancer in the MHB. This argues for the need to study different loci to find patterns that might help us predict what kind of logic is being used. It is also generally unclear how long functional enhancers are and what the minimum enhancer length is. While short enhancers are sufficient to drive transgenic expression, enhancers identified in the genome tend to be long, and predictions using chromatin marks also give us pretty large pieces. However, the results in the preprint suggests that the functional units of enhancers can be small, down to the scale of tens of bps, and that each important unit can be narrowed down to one or two TFBS. This argues that we might need to think about how we define enhancers in general, and that to understand enhancer biology, we not only need to study interactions and contribution of multiple enhancers to gene expression, we also need to understand how TFBS interact to decipher each functional unit. I also find it interesting that there is redundancy and cooperation observed at the length scales of multiple TFBS (in the case of CE64) and at the length scales of multiple enhancers (in the case of the limb). Finally, I really like the idea of functional units being conserved but rewired for different regulatory logic.
Future directions and questions
- Given that expression level and phenotype were both characterised in this study, is there some expression level cutoff at which phenotypic defects are observed? If so, how much Fgf8 does the mice have to lose to lead to a phenotypic change?
- This might not be easy to do, but is there a way to look at ChIP or other data to see if the predicted TFBSs are bound by the TF? Also, is mutation of the TFBS sufficient to abrogate CE64 function?
- It would be interesting to look at double deletions of enhancers in the limb AER to see if all four enhancers are redundant for each other or some other case, for example, if there are perhaps two groups of two redundant enhancers and one from each group is necessary.
doi: https://doi.org/10.1242/prelights.17824
Read preprint (1 votes)
(1 votes)
5 years
Jawaher
Hi,
I am a PhD student. Can I contact you to discuss how to interpret some of my data regarding conserved TFBS motifs, please?