








Inference of drug off-target effects on cellular signaling using Interactome-Based Deep Learning
Posted on: 29 November 2023 , updated on: 7 May 2024
Preprint posted on 15 October 2023
Article now published in iScience at http://dx.doi.org/10.1016/j.isci.2024.109509
Unleashing artificial brain power to play matchmaker for drugs and cellular signalling – because even diseased cells deserve a perfect match!
Selected by Benjamin Dominik MaierCategories: bioinformatics
Updated 29 April 2024 with a postLight by Benjamin Dominik Maier
Congratulations to Nikolaos Meimetis, Douglas A. Lauffenburger and Avlant Nilsson! Their preprint has now been published in iScience (link to the article). Although the manuscript remained largely the same, notable changes include an expanded lethality analysis and the addition of a new section exploring prediction performance across drug classes further improving the already worth-reading article.
While the authors only trained a random forest model to predict lethality in the preprint, 10 regularisation, regression and classification methods have been trained for the journal version. All methods were shown to outperform randomised models. Based on all their findings, the authors now conclude that the experimentally observed lethality can be fully explained with just the on-target effects without the need to include inferred interactions. Additionally, the authors have identified a known therapeutical target, MAPK12 kinase, which can explain lethality without being affected by the tested drugs (off-target effect).
Building up on their Lestaurtinib case study, the authors analysed more FMS-like tyrosine kinase 3 (FLT3) inhibiting drugs with their mechanism of action inference method. Their new results indicate that the model might be able to find underlying trends within specific drug classes (model generalisation). They further explored this by evaluating the models’ predictions across whole drug classes and conclude that a) the model performance and inferred off-targets vary between different classes of drugs (new Figure 5) and b) their approach may help to inform experimental design.
In the discussion section, the authors added a paragraph outlining the limitations of their study and suggest potential follow-up analysis to further improve their model performance and interpretability. Newly added supplementary figures further help to understand their method and provide more in-depth results for the interested reader.
Background
Cellular Signalling
Biological signalling is an essential property of living organisms allowing cellular communication and coordinated responses to external environmental changes. Signalling is initiated by signalling molecules binding to receptors on the cell surface (reception), leading to a conformational change of the receptor. This results in a series of intracellular events (e.g. phosphorylation cascades), which amplifies and relays the signal through the cell (signal transduction). The transduced signal ultimately leads to a specific cellular response such as changes in gene expression and alterations in enzyme activity.
Diseases like cancer (Sever & Brugge, 2015), diabetes (Onyango, 2020) and others (Spiegel, 1996 & Zhang et al., 2019) can result from cell signalling deregulation, which is usually caused by genetic mutations, environmental factors, or a combination of both. Various therapeutic approaches are being explored to either restore cellular balance or eliminate diseased cells. One strategy to restore normal cellular behaviour involves the use of small molecules like kinase inhibitors (Zhong et al., 2021), which aims at suppressing kinases that are vital for tumour cell survival and proliferation while being less cytotoxic to non-cancerous cells. Another approach is the use of biologics, particularly monoclonal antibodies (Zahavi & Weiner, 2020). These specialised proteins bind to specific molecules involved in aberrant signalling pathways with high specificity to either block their activity or mark them for destruction by the immune system.
Artificial neural networks
Inspired by the way our brain works, an artificial neural network (ANN) is an interconnected group of nodes (“virtual neurons”) in a layered structure that are used for tasks like recognising patterns in images or making decisions based on data. ANNs consist of one input layer, multiple hidden layers and one output layer (Fig. 1). Similar to neurons, each node takes inputs from the previous layer, performs a calculation, and passes its output to the next node. Node connections are given numerical weights (w), with positive weights indicating that one node enhances the activity of another and negative weights indicating suppression. Nodes with higher weight values exert greater influence on the other nodes in the network. By learning from examples (training data), ANNs adjust the weights of these connections and thereby improve their performance over time.
The training data for many machine learning methods results from automated high-throughput screens like the Next Generation L1000 Connectivity Map (Subramanian et al., 2017) and/or the JUMP Cell Painting project (Chandrasekaran et al., 2023) which allow to a) identify drug mechanisms of action, b) nominate therapeutics for a particular disease, and c) construct biological networks among perturbations and genes. If you are interested in a similar approach where these datasets are being used, please check out our previous preLight post.
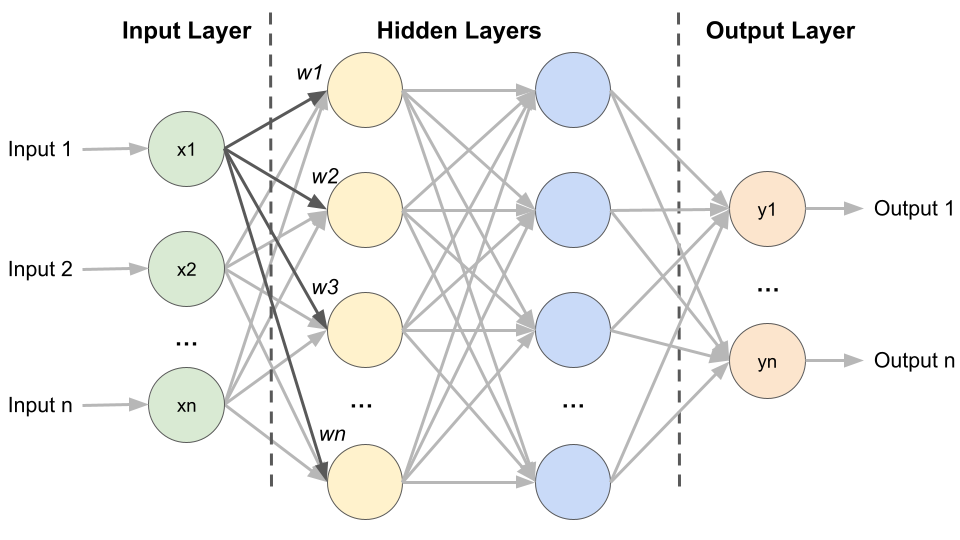
Fig. 1 Architecture of an Artificial Neural network. Created by Benjamin Maier in 2023.
Key Findings
Overview
Building upon their previously published Large-scale knowledge-EMBedded Artificial Signaling-networks (LEMBAS) (Nilsson, Peters et al., 2022), Meimetis and colleagues developed a computational model called DT-LEMBAS. Their new model uses artificial neural networks (ANN) to simultaneously predict drug-target interactions and their cell-line specific effects on intracellular signalling pathways.
The drug-target (DT) submodule of DT-LEMBAS takes the drug concentration as input to predict the signalling effects of the drugs on their targets. This is done using prior information from known drug-target interactions as well as chemical similarities between different drugs. The second module, LEMBAS, is a recurrent ANN model designed to simulate intracellular signalling processes. It takes the signalling effects of drugs as input to estimate the activity of transcription factors (TFs).
Unlike other approaches, DT-LEMBAS incorporates a prior knowledge network of signal transduction, along with information about transcriptional regulation and the chemical structure similarity of drugs. This enhances the interpretability of both on- and off-target effects on signalling cascades and ensures that the model is mechanistically plausible. The model outperforms conventional machine learning methods in predicting TF’ activity and identifies both known and novel drug-target interactions, which the preprint authors validated in independent datasets. As a case study (not featured in this preLight article), the authors applied DT-LEMBAS to analyse the impact of the drug Lestaurtinib on downstream signalling pathways, highlighting its potential to improve therapeutic design by identifying potential off-target drug effects.
Key Finding I: Benchmarking predictability of TF activities
First, the authors trained their model using the L1000 dataset (Subramanian et al., 2017) to identify TFs that could be predicted with high accuracy and validate whether the drug module predictions generalise using a cross-validation approach. They determined that their model accurately predicts the activities of many TFs across a wide range of cell lines. Notably, TFs showing strong fits in training also excelled in validation, while those with poor fits had lower performance, possibly due to noisy data or limited impact of the TF on the cell’s transcriptomic profile. Next, the model performance was benchmarked against four basic machine learning techniques. DT-LEMBAS consistently outperformed the other approaches, as well as the randomised models, as determined by statistical tests.
Key Finding II: Controlling the number of inferred interactions
To address gaps in drug-target interaction knowledge, the model must be able to infer new interactions. One approach is zero regularisation, allowing parameter adjustments without penalties thereby allowing drug connections with all potential targets (risk of overfitting). Conversely, infinite regularisation limits the model to known interactions (risk of under-fitting). Striking a balance, the authors employ L2 regularisation (Ridge regression) to add penalties as model complexity increases. This enables the model to infer drug-target interactions that are not part of the prior knowledge to better explain transcriptional data while preventing overfitting by restricting its ability to connect drugs with all targets.
The approach assigns importance scores to interactions, where negative indicates potential inhibition and positive suggests activation. Interactions with low scores are less relevant for explaining TF activity, while high scores are deemed important. Based on quantifying the impact of interaction removal on model accuracy for each drug, the authors chose a cutoff at a 25% increase in global error. This threshold successfully retrieved about 95% (670 out of 710) of known drug-target interactions, aligning with expectations, as these interactions are supported by literature or clinical evidence and are anticipated to contribute significantly to the observed transcriptional profile.
Key Finding III: Evaluating the inference of direct interactions by an ensemble of models
In addition to retrieving known interactions, the previous analysis found 13,000 novel interactions, which might represent undiscovered direct on-target drug-target interactions, indirect effects or model artefacts (false positives). Validating these predicted interactions is challenging due to the absence of ground truth data.
Instead, the authors made use of an independent database DrugBank which contains known direct drug-target interactions which were not present in the original model’s prior knowledge. They then attempted to predict these interactions based on how frequently they were inferred by their cell-line specific models as well as by some additional statistical metrics such as recall and F1 score. When considering all interactions that appeared in at least one model, the authors observed low precision (proportion of correctly predicted positive cases) and overall accuracy (proportion of all cases correctly classified). However, when an interaction was inferred more frequently, both accuracy and precision improved, reaching perfect precision for the most frequent predictions. This suggests that interactions found in multiple models are more likely to be direct interactions, which can be used for inferring novel drug-target interactions.
Key Finding IV: Identification of transcription factors regulated by off-target effects
Using their newly established criterion to predict direct on-target drug-target interactions, the authors identified off-target effects that contribute to the transcriptional profile. They removed all non-direct interactions and checked if the model could no longer explain the transcriptional profile, suggesting the presence of additional contributing interactions induced by off-target effects.
Case Study: A subnetwork explaining off-target effects of Lestaurtinib
Next, the authors explored (off-target) interactions for the tyrosine kinase inhibitor Lestaurtinib. The model identified an average of 82 potential interactions per model, including all known interactions found in the training dataset and in the DrugBank validation dataset. The interactions were then used to construct a subnetwork to elucidate Lestaurtinib’s mechanism of action on FOXM1. The resulting signalling network (Fig. 2) suggested that Lestaurtinib might cause an indirect inhibition of FOXM1 through either CDK1 and CDK2, which is supported by the literature. More detailed downstream analysis further supported the inhibition of FOXM1 through CDK2. Overall, the case study outlines how DT-LEMBAS combined with downstream validation analysis might be used to propose potential therapeutic interventions and drug combinations.
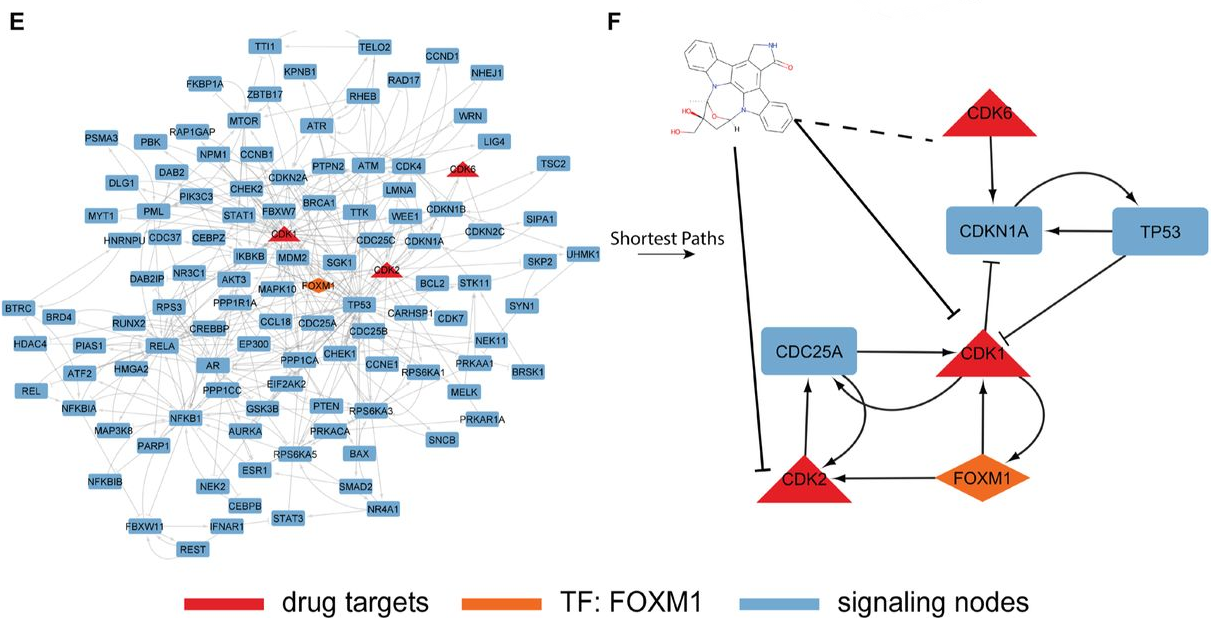
Fig. 2 Signalling network showing how Lestaurtinib inhibits FOXM1 through CDK1 and CDK2. Figure taken from Meimetis et al. (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Conclusion and Perspective
Despite significant reductions in the cost of computing and sequencing driven by technological advancements and economies of scale, the price per new drug has experienced an exponential increase over the past few decades. Nevertheless, given the exponentially increasing rate of data collection paired with advances in computational biology making use of the big data, there is a massive potential to accelerate drug design while reducing the associated costs and resources. For instance, virtual pre-screenings of large chemical libraries can narrow down the number of compounds that need to be experimentally tested to identify potential drug candidates. Moreover, as shown e.g. in the presented preprint, computational methods can identify potential drug targets by analysing biological data to understand the roles of specific proteins, genes, or pathways in diseases.
References
Nilsson, A., Peters, J.M., Meimetis, N. et al. Artificial neural networks enable genome-scale simulations of intracellular signaling. Nat Commun 13, 3069 (2022). https://doi.org/10.1038/s41467-022-30684-y
Onyango A. N. (2020). Mechanisms of the Regulation and Dysregulation of Glucagon Secretion. Oxidative medicine and cellular longevity, 2020, 3089139. https://doi.org/10.1155/2020/3089139
Sever, R., & Brugge, J. S. (2015). Signal transduction in cancer. Cold Spring Harbor perspectives in medicine, 5(4), a006098. https://doi.org/10.1101/cshperspect.a006098
Spiegel A. M. (1996). Defects in G protein-coupled signal transduction in human disease. Annual review of physiology, 58, 143–170. https://doi.org/10.1146/annurev.ph.58.030196.001043
Zahavi, D., & Weiner, L. (2020). Monoclonal Antibodies in Cancer Therapy. Antibodies (Basel, Switzerland), 9(3), 34. https://doi.org/10.3390/antib9030034
Zhang, L., Yousefzadeh, M. J., Suh, Y., Niedernhofer, L. J., & Robbins, P. D. (2019). Signal Transduction, Ageing and Disease. Sub-cellular biochemistry, 91, 227–247. https://doi.org/10.1007/978-981-13-3681-2_9
Zhong, L., Li, Y., Xiong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Sig Transduct Target Ther 6, 201 (2021). https://doi.org/10.1038/s41392-021-00572-w
doi: https://doi.org/10.1242/prelights.36027
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
The lipidomic architecture of the mouse brain
CRM UoE Journal Club et al.
Kosmos: An AI Scientist for Autonomous Discovery
Roberto Amadio et al.
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |