








Similarity metric learning on perturbational datasets improves functional identification of perturbations
Posted on: 17 August 2023
Preprint posted on 11 June 2023
Weak supervision - Strong results! Smith and colleagues introduce Perturbational Metric Learning (PeML), a weakly supervised similarity metric learning method to extract biological relationships from noisy high-throughput perturbational datasets
Selected by Benjamin Dominik Maier, Anna Foix RomeroCategories: bioinformatics, molecular biology, systems biology
Background
Similarity metric learning
Similarity metric learning is a technique to measure how similar or different things are from each other. Well known traditional similarity functions include the Pearson and Spearman correlation for omics modalities (Urbanczyk-Wochniak et al., 2003) and Gene Set Enrichment Analysis (GSEA) for gene expression analysis (Subramanian et al., 2005).
Machine learning-based similarity metric learning works in a weakly supervised manner (Duffner et al., 2021). This means that the similarity metric learning doesn’t try to categorise things into classes making it suitable when labels are unknown or hard to obtain (Hernández-González et al. 2016). Instead, it integrates known similarities from repeated measurements to create a high-dimensional space (embedding) where similar things are grouped together. Thus, it learns how to determine if new, unseen examples belong to the same class or exhibit similarity.
In the context of biology, similarity metric learning has proven particularly valuable for analysing large biological datasets. Biological measurements, such as gene expression or cell morphology, are often complex, exhibiting multimodal characteristics, susceptibility to confounding factors, and cell-to-cell variability (Eling et al., 2019). This complexity makes interpretation challenging, especially with sparse single-cell data and low signal-to-noise ratios. However, employing a similarity function tailored to the specific dataset transforms the data into a meaningful context-specific representation, enabling us to identify patterns and relationships within the dataset. For instance, it may help us to identify the mechanism of action, which is the specific way a treatment or substance affects a biological system.
High-throughput perturbational datasets
Recent advances in cost-effective transcriptomics and image-based profiling technologies have made it possible to create extensive public datasets allowing researchers to study the effects of chemical or genetic perturbations on cells, in an automated high-throughput manner. Notably, the Next Generation L1000 Connectivity Map (Subramanian et al., 2017) and the JUMP Cell Painting project (Chandrasekaran et al., 2023), developed through collaborations between pharmaceutical companies and research institutes, contain cell profiles of cells exposed to more than 100,000 unique compounds and genetic manipulations. These collaborations provide a unique opportunity to explore genetic patterns and similarities to a) identify drug mechanisms of action, b) nominate therapeutics for a particular disease, and c) construct biological networks among perturbations and genes.
Key Findings
Overview
Smith and colleagues introduce PeML, a weakly supervised similarity metric learning method that transforms biological measurements into an intrinsic, dataset-specific basis. Thus, biological relationships and mechanisms can be extracted from noisy high-throughput perturbational datasets. To measure the performance of the new method, the authors use the L1000 dataset comprising gene expression signatures of compounds in cancer and immortalised cell lines, as well as the CDRP Cell Painting dataset containing cellular morphology and function data from a single cell line. The authors show that PeML maximises the discrimination of replicate signatures, improves recall in biological data and yields better prediction of compound mechanisms of action. Recall (also known as sensitivity or true positive rate) is calculated as the ratio of the true positive (TP) predictions to the total number of actual positive instances in the dataset. PeML is capable of being learned with moderate dataset sizes and goes beyond traditional approaches by capturing a more profound notion of similarity. Therefore, it might improve data classification, clustering, and subsequent analyses.
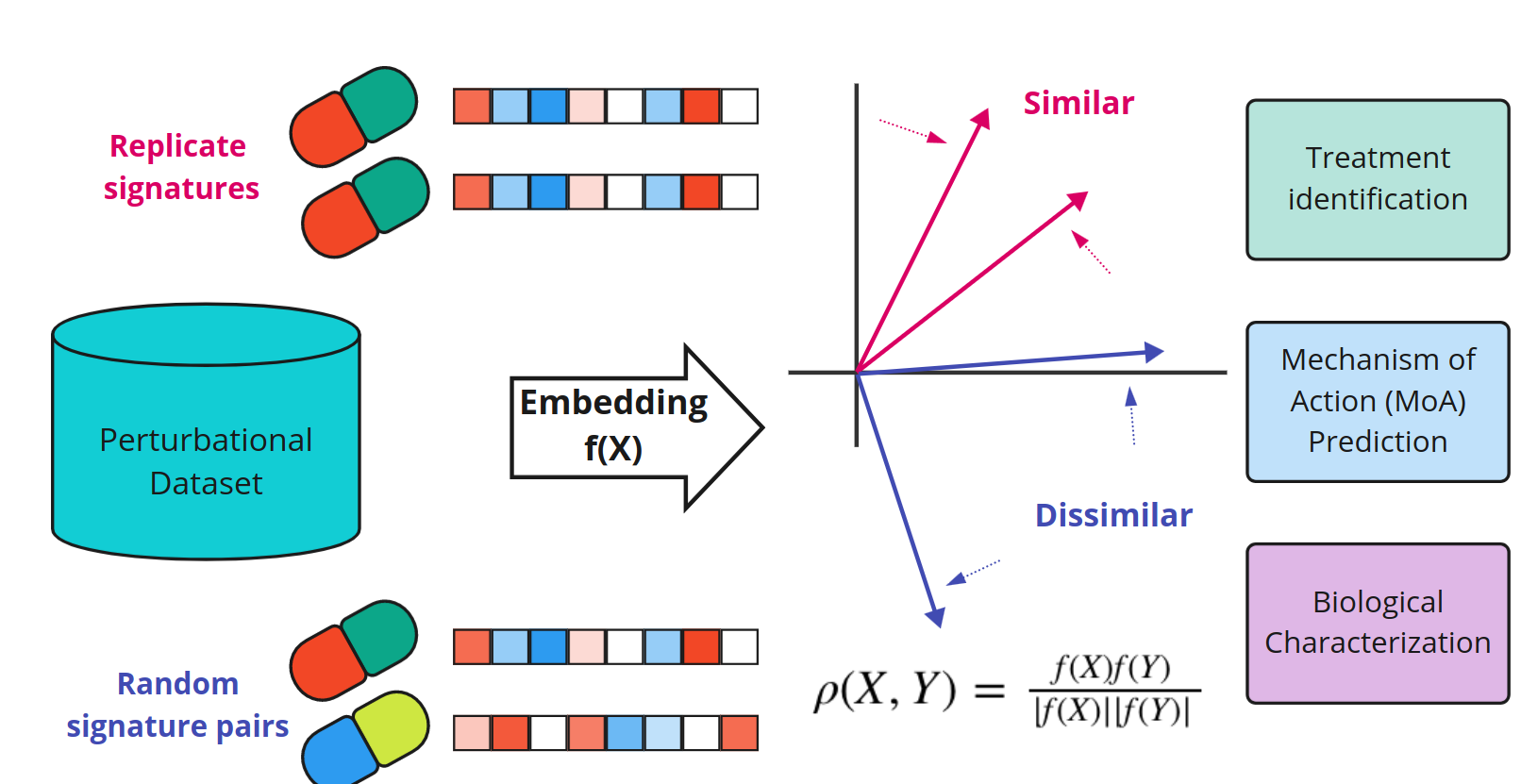
Fig. 1 Schematic of the weakly supervised ML similarity metric learning method Perturbational Metric Learning (PeML). Figure taken from Smith et al. (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Perturbational Metric Learning (PeML)
PeML is a weakly supervised machine learning framework that learns a similarity function between samples. This method uses replicates of experiments as ground truth to train a data-driven similarity function. Unlike traditional methods, PeML is a feature transformation technique that works directly on processed genetic or physical characteristics data, eliminating the need to extract new features from the original raw data.
PeML improves replicate recall in biological data
First, the authors conducted a replicate recall analysis to quantify the model’s ability to capture biologically relevant relationships in the data. To account for differences between cell lines, separate context-specific models were trained for each cell line. The training was performed on small batches of data instead of the entire dataset at once (mini-batch stochastic gradient descent), making it more efficient. Signatures representing the same compound treatment were grouped together, regardless of dosage or time point. As a similarity metric balanced AUC was used, which adjusts for some classes having more examples than others. AUC is a metric for evaluating machine learning models in binary classification tasks. It measures the area under the Receiver Operating Characteristic curve, where TPR (correctly classified positive samples) is plotted against FPR (incorrectly classified negative samples), providing insight into the model’s ability to distinguish between classes.
Subsequently, the model’s generalizability and performance across different compounds was evaluated using 5-fold compound-wise cross-validations. This means that the dataset was split into five parts based on the compounds, and each part was used as a validation set once while the other four parts were used for training. Thus, the authors demonstrated that PeML outperformed the baseline cosine similarity, yielding higher replicate rank and improving recall for replicate pairs in various cell lines, as well as achieving better results for previously unseen compounds.
PeML improves prediction of compound mechanism of action from perturbational signatures
Next, the authors benchmarked PeML’s ability to identify drugs’ mechanisms of action. Across each cell line in the L1000 and Cell Painting datasets, they found that PeML recovers a greater proportion of biologically-relevant mechanisms of action. Furthermore, a signal-to-noise ratio analysis revealed that PeML better discriminates similar pairs from the background than standard similarity metrics.
Generalizability of PeML
While the previous analyses demonstrated promising results for large high-quality datasets, the performance on smaller training datasets remained unknown. Hence, the authors assessed the minimal training data required for a well-generalised model by downsampling the original datasets. Their results indicate that a few hundred conditions with replicates are sufficient to identify and retrieve biologically relevant associations from a given dataset.
Finally, the authors tested their initial hypothesis that context-specific models tailored to a specific cell line perform better than pan-models trained on all cell lines. The results demonstrated that learning context-specific models for different cancer cell lines improved similarity retrieval tasks compared to models trained in all contexts and cosine models.
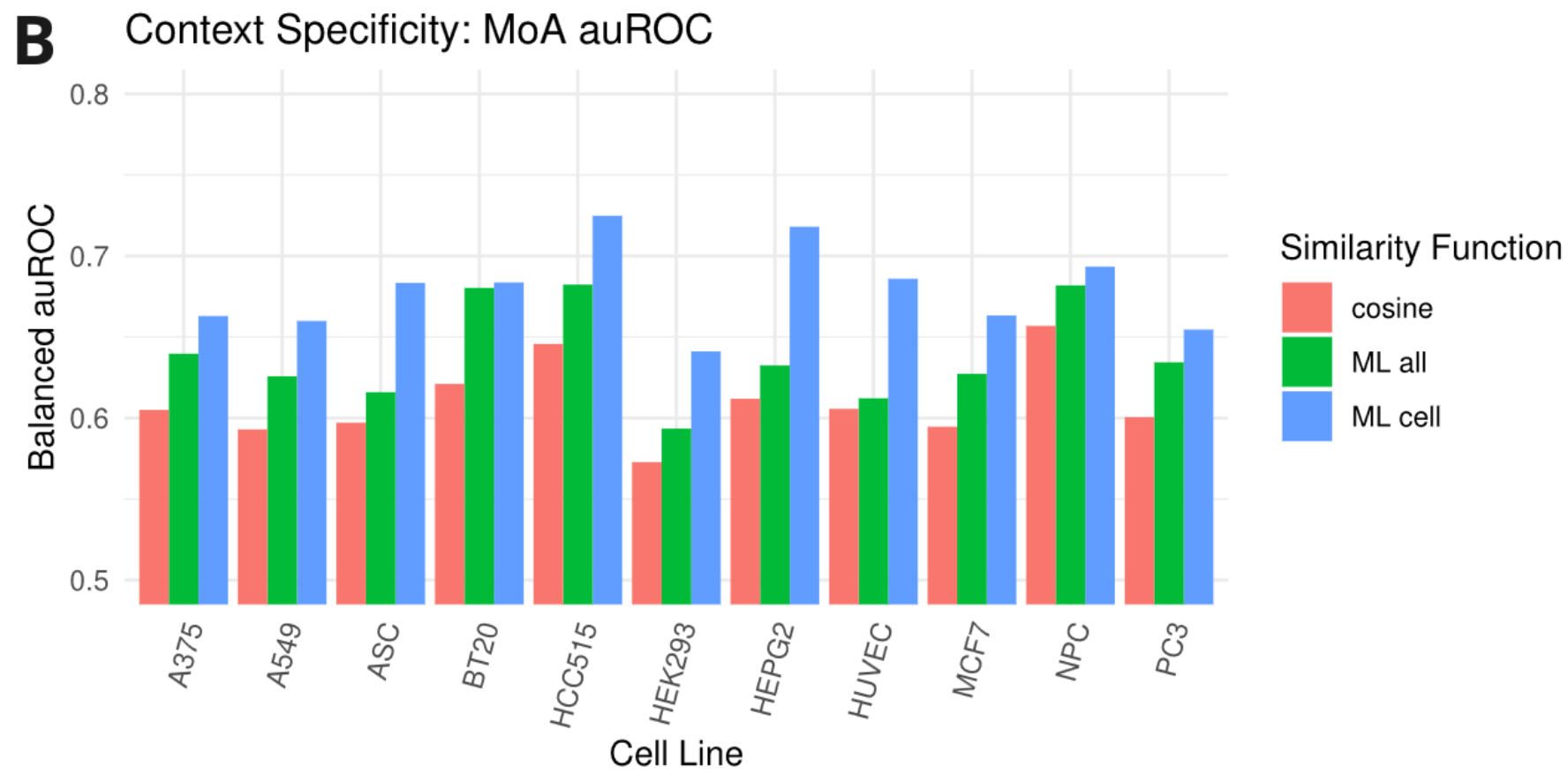
Fig. 2 Cell line-specific metric learning functions outperform a pan-dataset function and a baseline cosine function in predicting Mechanism of Action. Figure taken from Smith et al. (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International license.
Further Material
R package (not released yet)
Conclusion and Perspective
As the volume of large-scale biological datasets continues to grow, the increasing relevance of weakly supervised learning algorithms becomes evident, offering data-driven and scalable analysis while minimising the dependency on costly and time-consuming expert annotations and training data. In this preprint, Smith and colleagues present Perturbational Metric Learning (PeML), a powerful tool for the analysis of large biological datasets. PeML learns a data-driven similarity function by transforming biological measurements into an intrinsic, dataset-specific basis to extract meaningful biological associations such as compound mechanisms of action from noisy datasets. In addition to capturing a more meaningful notion of similarity, data in the transformed basis can be used for other analysis tasks, such as classification and clustering.
The idea of integrating large-scale imaging data into our pipelines has emerged as a pressing challenge. This led us to consider featuring a preprint that offers valuable insights into bridging multi-omics data analysis with imaging and machine learning. Given Benjamin’s expertise in integrating and analysing large-scale multi-omics data, along with Anna’s background in computer vision, bioimage analysis and machine learning, this preLight post presents an exciting opportunity for interdisciplinary collaboration.
References
Chandrasekaran, S. N., Ackerman, J., Alix, E., Ando, D. M., Arevalo, J., Bennion, M., Boisseau, N., Borowa, A., Boyd, J. D., Brino, L., Byrne, P. J., Ceulemans, H., Ch’ng, C., Cimini, B. A., Clevert, D.-A., Deflaux, N., Doench, J. G., Dorval, T., Doyonnas, R., … & Carpenter, A. E. (2023). JUMP Cell Painting dataset: morphological impact of 136,000 chemical and genetic perturbations. bioRxiv. https://doi.org/10.1101/2023.03.23.534023
Eling, N., Morgan, M. D., & Marioni, J. C. (2019). Challenges in measuring and understanding biological noise. Nature reviews. Genetics, 20(9), 536–548. https://doi.org/10.1038/s41576-019-0130-6
Hernández-González, J., Inza, I., & Lozano, J. A. (2016). Weak supervision and other non-standard classification problems: A taxonomy. Pattern Recognition Letters, 69, 49–55. https://doi:10.1016/j.patrec.2015.10.008
Stefan Duffner, Christophe Garcia, Khalid Idrissi, Atilla Baskurt. Similarity Metric Learning. Multi-faceted Deep Learning – Models and Data, 2021. ⟨hal-03465119⟩ https://hal.science/hal-03465119
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., … & Mesirov, J. P. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences, 102(43), 15545-15550. https://doi.org/10.1073/pnas.0506580102
Subramanian, A., Narayan, R., Corsello, S. M., Peck, D. D., Natoli, T. E., Lu, X., Gould, J., Davis, J. F., Tubelli, A. A., Asiedu, J. K., Lahr, D. L., Hirschman, J. E., Liu, Z., Donahue, M., Julian, B., Khan, M., Wadden, D., Smith, I. C., Lam, D., Liberzon, A., … & Golub, T. R. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell, 171(6), 1437–1452.e17. https://doi.org/10.1016/j.cell.2017.10.049
Urbanczyk-Wochniak, E., Luedemann, A., Kopka, J., Selbig, J., Roessner-Tunali, U., Willmitzer, L. and Fernie, A.R. (2003), Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO reports, 4: 989-993. https://doi.org/10.1038/sj.embor.embor944
doi: https://doi.org/10.1242/prelights.35232
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the molecular biology category:
Defective BRCA1-mediated DNA end resection drives tandem duplication formation and FANCM synthetic lethality
Marta San Martin
Thousandfold Expansion Microscopy
Felipe Del Valle Batalla
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Also in the systems biology category:
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |