








Inferring protein from mRNA concentrations using convolutional neural networks
Posted on: 20 December 2023 , updated on: 31 March 2025
Preprint posted on 6 November 2023
Article now published in BioData Mining at https://doi.org/10.1186/s13040-025-00434-z
Convolutional Gene Expression Detective: Decoding mRNA Features to Predict Protein Abundance
Selected by Benjamin Dominik MaierCategories: bioinformatics
Updated 31 March 2025 with a postLight by Benjamin Maier
Congratulations to Patrick Maximilian Schwehn and Pascal Falter-Braun on the publication of their manuscript in BioData Mining! While the manuscript closely mirrors the preprint, significant improvements have been made in the final version. The text has been thoroughly rewritten and restructured, enhancing the clarity of the results and conclusions, and contributing to better reproducibility (e.g., by including package versions).
A new methods section has been added, providing a detailed explanation of the cross-correlation approach. Additionally, Figures 2 and 3 have been updated with new panels: Figure 2C now includes the learned weights for amino acid usage in the single-feature model, and Figure 3B presents a histogram of manually selected GO terms for potential regulatory genes.
Moreover, the authors tested whether changes in transcript abundance directly reflect protein abundance changes, ultimately concluding that normalizing protein abundances by transcript abundance changes is less effective than using average protein abundances alone.
Background
Regulation of Protein Abundance
Almost all biological functions, such as metabolism, signalling, transport, mechanical processes, and immune responses are governed by proteins. Hence, protein abundance undergoes rigorous regulation across various stages of synthesis and degradation (Fig. 1). Collectively these highly redundant mechanisms regulate the cell’s protein abundance ensuring stability while retaining adaptability to changing conditions.
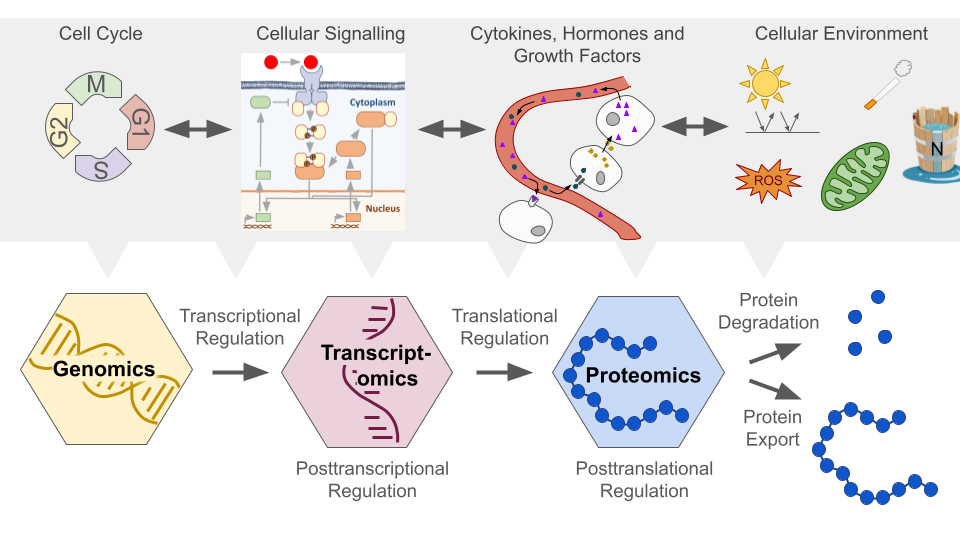
Fig. 1 Regulation of Protein Abundance. Figure created by Benjamin Maier (2023) in PowerPoint. At the transcriptional level, gene expression is modulated by sequence motifs and regulatory elements such as promoters, enhancers, transcription factors, and epigenetic modifications. Post-transcriptionally, RNA processing (splicing, capping and poly-adenylating) affecting mRNA stability and gene silencing by RNA interference regulate mRNA levels. Translation itself is mainly regulated through initiation factors and codon availability. Post-translationally, protein folding, modifications like phosphorylation and ubiquitination, as well as degradation via proteasomes and lysosomes and export are pivotal in determining protein levels. The cellular environment (e.g. stress responses), cellular signalling, cell cycle regulation, and external signals further contribute to this complex regulatory system.
Convolutional Neural Networks (CNNs)
Neural networks consist of interconnected layers of nodes. Nodes have weights and if a node’s output exceeds its threshold, data is transmitted to the next layer; otherwise, no information is forwarded. Convolutional neural networks are a specific type of neural network consisting of convolutional layer(s), pooling layer(s) and fully-connected layer(s) (Fig. 2). Check out my recent preLight post about Interactome-Based Deep Learning for more general information about neural networks.
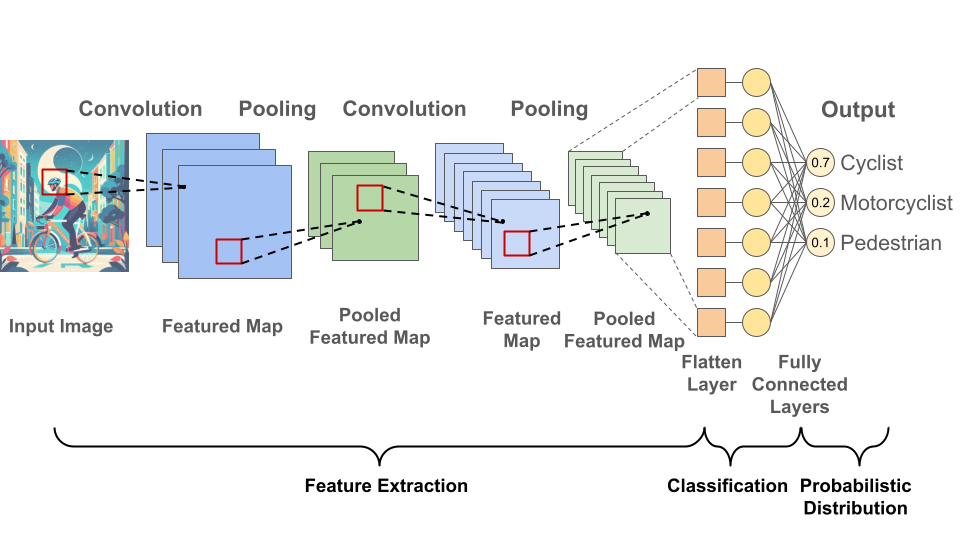
Fig. 2 Convolutional Neural Network Architecture. Figure created by Benjamin Maier (2023) in PowerPoint. The convolutional layers extract features like edges, textures, or shapes from the input data using sliding filters and conducting element-wise multiplications and additions. The pooling layers then condense the information obtained from the convolutional layer by reducing its spatial dimensions, which prevents overfitting and improves computational resource management. Finally, fully-connected layers perform the task of classification based on the features extracted through the previous layers and their different filters.
Key Findings
Overview
Understanding context-specific quantitative protein dynamics is crucial for comprehending and modelling molecular processes. While large-scale proteomic quantifications remain technically challenging and expensive, transcriptomics is comparatively cheap and widely available. However, there is only a low correlation between the protein species and their cognate mRNA transcripts for many proteins involved in protein translation, mRNA splicing, oxidative phosphorylation, electron transport chain, and other housekeeping processes (Komili & Silver (2008), Mertins et al. (2016), Zhang et al. (2016), Clark et al. (2019), Zhang et al. (2022), Srivastava et al., 2022).
Schwehn and Falter-Braun analysed matched transcriptome-proteome data from 29 human tissues (Wang et al., 2019) and 30 plant tissues (A. thaliana) (Mergner et al., 2020). Using a convolutional neural network approach with a linear regressor, they extracted the most meaningful mRNA features to infer protein levels from mRNA concentrations for multiple human and plant tissues. Their analysis determined that coding sequences-derived features are most predictive when trying to infer protein abundance. Immune related (humans) and environmental related processes were found to be associated with the strongest transcript-protein correlations. Overall, their predictions yielded better performance than earlier studies.
Key Finding I: Understanding transcript-protein relationships
After data preprocessing, the authors filtered out genes and proteins expressed in too few samples to subsequently run a regression analysis aimed at identifying the mathematical expression best describing the relationship between mRNA and protein levels. Even though more complex models like square root functions were tested, linear regression yielded the most accurate predictions, indicating potential overfitting of other models due to limited data.
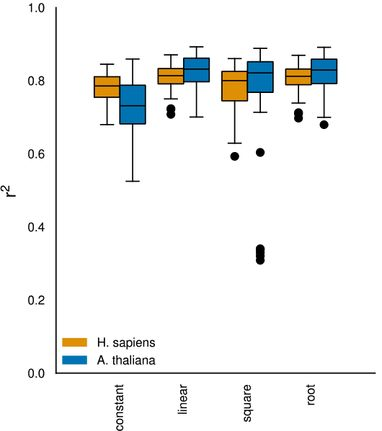
Fig. 3 Variation explained (coefficient of variation, R2 value) by various predictors in the regression model. Figure taken from Schwehn and Falter-Braun (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Next, the authors looked at the best and worst fitting genes in humans and A. thaliana. Although mRNA levels varied, approximately a quarter of the genes showed minimal to no alteration in their protein levels. Moreover, some genes were found with a negative correlation between transcript and protein levels indicating complex regulatory mechanisms. Overall, the authors observed a greater transcriptional variance in A. thaliana and higher protein-level variance in humans which suggests that A. thaliana relies more on transcriptional regulation while translational regulation is more important for humans.
Key Finding II: Linear regressor for predicting protein-to-RNA ratios
Subsequently, the authors employed linear regressors to predict transcript-to-protein relationships and identify which mRNA features have the strongest impact on protein level predictability. For fixed sequence features like codon counts, the authors used dense layers while convolutional layers were employed for sliding window sequence features such as the nucleotide coding sequence. Coding sequence-derived features, such as amino acid content and codon counts, yielded the highest predictive capability. Interestingly, out-of-frame codon counts performed comparably to in-frame ones, attributed by the authors to consistent underlying trends in amino acid composition and the low importance of the wobble base. Combining all sequence features slightly improved model performance but only marginally surpassed the most informative individual input feature, the peptide sequence, (Fig. 4), indicating a saturation of predictive power due to shared information/redundancy.
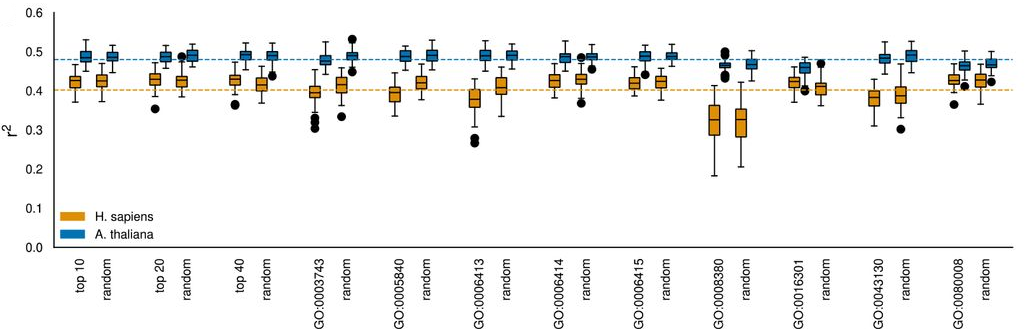
Fig. 4 Predictive power (coefficient of variation, R2 value) of various sequence input features. Figure taken from Schwehn and Falter-Braun (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Next, the authors quantified the impact of specific codons on protein abundance. In humans, high counts of charged amino acids were found to have the greatest positive impact on protein-to-RNA ratios. In contrast, Isoleucine displayed the highest predictability in Arabidopsis whereas no such effect could be observed in humans. Overall, they observed hydrophobic amino acids were more predictive for protein levels in A. thaliana, which is possibly linked to varying body temperatures and biophysical protein demands (37°C for humans, 4°C to 34°C for Arabidopsis). In both species, start-codon, stop-codons and their neighbourhoods were found to be highly informative.
Key Finding III: Analysis of cell-context specific correlations
Finally, the model was expanded to account for possible attractors meaning that the level of a certain protein might be modulated by the transcript of a related gene. Hence, the authors included their mRNA expression scores and relationships as proxies for protein expression scores across different gene sets to see whether this further improved the predictions. Two methods were used to define gene sets: a quantitative approach based on gene-gene correlations and a qualitative approach using gene ontology terms related to translation or protein decay.
The analysis revealed that certain gene sets, particularly those linked to immune responses in humans and environmental responses in Arabidopsis are key effectors and strongly correlate with protein expression levels. Yet, the study found that there was no considerable increase in the prediction performance when accounting for gene sets, instead the model often performed similar to randomly sampled genesets.
Conclusion and Perspectives
Despite recent advances in large-scale protein quantifications (e.g. protein sequencing) and paired multi-omics measurements (e.g. REAP-seq), whole-cell protein expression information will not be available for most experimental settings in (near) future. Machine learning-based methods to infer protein abundance can fill the gap by estimating protein abundance from readily available transcriptomics measurement. Moreover, by analysing the mRNA features found to be most informative, we can improve our limited understanding of protein regulation and its mechanisms.
When reading this preprint, I really liked the comparison between feature importance in humans and plants, because I only rarely see machine learning applications being used across species in a comparative way. Moreover, it was interesting to see how sequence features can help to improve prediction accuracy given that many researchers discussed its potential benefits in recent years. Still, the article left me wondering on whether combining it with other recent ML approaches like Li et. al, work from 2019 would improve the results considerably and how to better benchmark different ML predictors to get a better understanding on which estimator performs best where.
Questions to the Authors
Q1: How well do the best predicting genes and significantly enriched biological processes compare to previous machine learning studies such as Srivastava et al., 2022 or Li et. al, 2019?
Q2: I always struggle to compare different ML approaches to infer protein levels from mRNA concentrations given the lack of widespread standard benchmarking routines. How do you compare your methodology to other published approaches?
Q3: When reading about your analysis of cell-context specific correlations accounting for possible attractors, I became curious about the rationale behind the analysis design presented in your manuscript. I’m particularly interested in understanding how and why you chose this approach and how it compares to the gene-specific and trans-tissue models from Li et. al, 2019?
Q4: While I understand that all ML approaches have a slightly different aim (e.g. tuned for different cellular context) and settings in which they perform better than all competitors, I often observe a tendency toward the development of new methods rather than the refinement and enhancement of existing ones. This seems necessary within the current publishing system but appears highly inefficient to me. What are your thoughts on this and how could this practice be transformed?
Q5: Like most modern approaches, your ML model was trained with sequence features and experimental data from steady-state conditions. How well-suited do you believe these models are in predicting protein abundance from mRNA levels in dynamic, perturbed systems? What are the challenges that must be addressed to improve their performance in these dynamic scenarios?
References
Clark, D. J., Dhanasekaran, S. M., Petralia, F., Pan, J., Song, X., Hu, Y., da Veiga Leprevost, F., Reva, B., Lih, T. M., Chang, H. Y., Ma, W., Huang, C., Ricketts, C. J., Chen, L., Krek, A., Li, Y., Rykunov, D., Li, Q. K., Chen, L. S., Ozbek, U., … Clinical Proteomic Tumor Analysis Consortium (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell, 179(4), 964–983.e31. https://doi.org/10.1016/j.cell.2019.10.007
Komili, S., & Silver, P. A. (2008). Coupling and coordination in gene expression processes: a systems biology view. Nature Reviews Genetics, 9(1), 38–48. https://doi.org/10.1038/nrg2223
Li, H., Siddiqui, O., Zhang, H., & Guan, Y. (2019). Joint learning improves protein abundance prediction in cancers. BMC biology, 17(1), 107. https://doi.org/10.1186/s12915-019-0730-9
Mergner, J., Frejno, M., List, M., Papacek, M., Chen, X., Chaudhary, A., … Kuster, B. (2020). Mass-spectrometry-based draft of the Arabidopsis proteome. Nature, 579(7799), 409–414. https://doi.org/10.1038/s41586-020-2094-2
Mertins, P., Mani, D. R., Ruggles, K. V., Gillette, M. A., Clauser, K. R., Wang, P., … NCI CPTAC. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature, 534(7605), 55–62. https://doi.org/10.1038/nature18003
Srivastava, H., Lippincott, M. J., Currie, J., Canfield, R., Lam, M. P. Y., & Lau, E. (2022). Protein prediction models support widespread post-transcriptional regulation of protein abundance by interacting partners. PLOS Computational Biology, 18(11), 1–27. https://doi.org/10.1371/journal.pcbi.1010702
Wang, D., Eraslan, B., Wieland, T., Hallström, B., Hopf, T., Zolg, D. P., Zecha, J., Asplund, A., Li, L. H., Meng, C., Frejno, M., Schmidt, T., Schnatbaum, K., Wilhelm, M., Ponten, F., Uhlen, M., Gagneur, J., Hahne, H., & Kuster, B. (2019). A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Molecular systems biology, 15(2), e8503. https://doi.org/10.15252/msb.20188503
Zhang, H., Liu, T., Zhang, Z., Payne, S. H., Zhang, B., McDermott, J. E., … Townsend, R. R. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell, 166(3), 755–765. https://doi.org/10.1016/j.cell.2016.05.069
Zhang, Y., Chen, F., Chandrashekar, D. S., Varambally, S., & Creighton, C. J. (2022). Proteogenomic characterization of 2002 human cancers reveals pan-cancer molecular subtypes and associated pathways. Nature Communications, 13(1), 2669. https://doi.org/10.1038/s41467-022-30342-3
doi: https://doi.org/10.1242/prelights.36098
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
The lipidomic architecture of the mouse brain
CRM UoE Journal Club et al.
Kosmos: An AI Scientist for Autonomous Discovery
Roberto Amadio et al.
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |