








Inter-organ growth coordination is mediated by the Xrp1/Dilp8 axis in Drosophila
Posted on: 6 January 2019
Preprint posted on 5 November 2018
Article now published in Developmental Cell at http://dx.doi.org/10.1016/j.devcel.2019.03.016
One gene to rule them all: local growth defects caused by impaired ribosomal activity in the fly wing disc trigger systemic growth reduction via Xrp1-mediated activation of Dilp8
Selected by Alberto Rosello-DiezBackground
The regulation of organ size during development and repair is one of the last frontiers of biology, as we lack a fundamental understanding of whether/how organ size is encoded and sensed, and how it is fine-tuned to compensate for developmental noise or genetic and environmental perturbations. This compensation is particularly challenging if only part of the organism is affected (local perturbation), as body proportions have to be maintained/restored.
In the last decade, a few groups have capitalised on the power of Drosophila genetics to study this topic, and found that upon local perturbation in larval tissues, damaged imaginal discs secrete an alarm signal (Dilp8) that eventually leads to reduced levels of the hormone ecdysone, with two major consequences: a) delayed metamorphosis and b) systemic growth reduction, helping the damaged tissues to catch-up (1-4). Interestingly, Dilp8 seems to be the master gene of inter-organ communication, as different types of insults (developmental noise, cell death, impaired ribosomal activity) converge on Dilp8 activation, Despite the relevance of these findings, an obvious outstanding question is how the damage or the defective growth is detected and ends up activating secretion of Dilp8. Here, Boulan et al. explore the role of Dilp8 in the systemic growth reduction triggered by impaired growth in the wing pouch, and manage to find its upstream activator. For that, they went back to the original screen that revealed dilp8 role in the developmental delay triggered by impaired imaginal disc growth (1) and explore the role of one candidate that did not play a major role in the developmental delay caused by neoplastic growth, but seems to do so in the systemic growth reduction caused by ribosomal deficiencies.
Key findings
The authors knocked down the ribosomal protein RpL7 specifically in the wing pouch, causing cell-autonomous growth impairment as compared to control flies (“minute” discs hereafter). They then characterise intra-organ and inter-organ proportions and report that the growth defect in the minute wing pouch is accompanied by cell-nonautonomous reduction in other regions of the wing disc (hinge and notum), and also in distant discs such as the eye one. Similar effects are obtained by silencing RpS3. An exciting “hunt” then begins to elucidate the proximate and ultimate causes of this intra- and inter-organ coordination. These are their main findings:
- RNAi-mediated inhibition of dilp8 in the pouch fully rescues the growth defect in the hinge and notum, and in the eye disc, but not in the wing pouch, leading to perturbed intra- and inter-organ proportions.
- dilp8 overexpression in the wing pouch is enough to trigger the growth defect in the hinge and notum and in the eye disc, but not in the pouch itself, confirming the cell-nonautonomous role of Dilp8 in the growth defects.
- Inhibition of the previously known activators of dilp8 expression (JNK and Hippo signalling pathways) does not preclude the upregulation of dilp8 nor does it rescue the inter-tissue growth coordination, revealing that a different pathway is involved in the response to ribosomal deficiencies.
- xrp1, encoding a bZIP transcription factor and one of the candidates that came out of the original screen for developmental delay causers (1), seems to do the trick. When rp1 is silenced in the minute pouch, dilp8 expression is not triggered, and the intra- and inter-organ growth coordination are rescued, whereas the cell-autonomous growth defect is not.
- xrp1 overexpression in the pouch causes two effects: a) local apoptosis and growth reduction, and b) non-autonomous growth inhibition. The latter effect is rescued by silencing dilp8, while the former is not, suggesting Dilp8 only mediates the cell-nonautonomous effects of Xrp1.
- xrp1 levels are only modestly increased in slowly-growing wing pouches, suggesting that subtle transcriptional changes are enough to trigger the downstream effects, or that subsequent post-translational modifications are also required.
- Regarding upstream regulators of Xrp1, p53 and oxidative stress were previously shown to activate xrp1. However, Boulan et al. show that inhibition of these pathways in minute discs does not impede the non-autonomous growth defect. Xrp1 was recently shown to be involved in the phenomenon of cell competition that leads to elimination of minute clones when confronted to WT cells (5), and thus the authors explore other key players of this phenomenon, such as the ribosomal protein RpS12. This protein turns out to be a crucial player, because while its silencing causes a strong cell-autonomous growth inhibition in the pouch, it does not lead to the non-autonomous effects that the downregulation of RpL7 or RpS3 do. This strongly suggests that RpS12 is a key part of the sensor that translates growth defects into the Xrp1/Dilp8 response. See also the Figure below.
- Indeed, downregulation of RpS12 in the minute wing pouch precludes dilp8 upregulation and rescues the non-autonomous growth defect.
In conclusion, this study reveals yet another upstream regulator of dilp8, adding to the idea that multiple types of lesions converge, via different pathways, into a master coordinator of systemic growth. Truly one ring gene to rule them all?
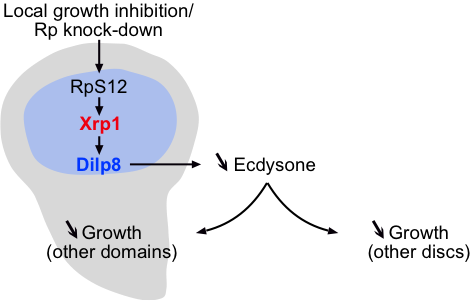
Figure. Schematic showing the key players found involved in inter-organ communication
What I like about this preprint
Using relatively simple but very elegant experiments, this study manages to go one step further and add new players to the fascinating and intriguing field of growth coordination. By revealing that different lesions converge on dilp8 activation via different routes, this study will likely be a guiding lighthouse for others navigating the dark waters of this field. From my own professional perspective, as a vertebrate developmental biologist studying the same topic, this study (and the previous related ones) is truly inspirational.
Pending questions
- Is comparison with other discs necessary? In other words, if all discs were similarly affected, would xrp1 still be activated? From an evolutionary point of view, it could be argued that systemic growth reduction does not bear any selective advantage when all imaginal tissues are equally affected.
- Is Xrp1 triggered upon physical lesion in the disc, or only by ribosomal defects? If Xrp1 is indeed activated in other types of lesions, especially those that require local compensatory proliferation, is Xrp1 involved in those local repair mechanisms?
- Inhibition of JNK or Hippo signalling does not rescue dilp8 upregulation in this context, but the authors mention it does partially rescue the developmental delay at the time of metamorphosis. Is this effect Dilp8-indepependent, or is Dilp8 expression/activity eventually affected by the inhibition of these signalling pathways?
- What happens if local growth is promoted instead of inhibited? The authors previously showed that tumours also trigger Dilp8-mediated developmental delay, but what about systemic growth reduction, especially as they now show that xrp1 is not upregulated?
- Moreover, tumorigenic lesions often end up in activation of the JNK signalling pathway, probably explaining the activation of Dilp8. What happens if a less “conspicuous” overgrowth is triggered? Do other tissues overgrow such that body proportions are maintained?
- The authors show that inhibition of p53 or oxidative stress does not rescue the non-autonomous growth effect induced by minute discs, but what about xrp1 levels? If they were indeed changed, that would be a strong argument in favour of post-translational modifications being the key regulator of Xrp1 activity upon local growth impairment.
- An obvious one: how does RpS12 detect the growth defect? Does it have to do with the way the growth defect is triggered (i.e. affecting also ribosomal proteins?). Is RpS12 also involved if the defect is triggered by blocking the cell cycle, instead of affecting ribosomal activity?
Related research
- J. Colombani, D. S. Andersen et al. Science 336, 582-585 (2012).
- A. Garelli, A. M. Gontijo et al. Science 336, 579-582 (2012).
- J. S. Jaszczak, J. B. Wolpe et al. Genetics 204, 703-709 (2016).
- J. S. Jaszczak, J. B. Wolpe et al. Genetics 200, 1219-1228 (2015).
- L. Baillon, F. Germani et al. Sci Rep 8, 17712 (2018).
doi: https://doi.org/10.1242/prelights.6943
Read preprint (No Ratings Yet)
(No Ratings Yet)