








RAD50 promotes DNA repair by homologous recombination and restrains antigenic variation in African trypanosomes
Posted on: 22 April 2020
Preprint posted on 17 March 2020
Article now published in Nucleic Acids Research at http://dx.doi.org/10.1093/nar/gkaa1265
Categories: microbiology
Background:
In humans, ‘Sleeping Sickness’ (African Trypanosomiasis) is an often fatal, neglected tropical infection caused by the parasite Trypanosoma brucei. T. brucei lives in skin, fat tissues and blood meaning they are in constant contact with the host immune system. To avoid clearance, T. brucei parasites are covered in a thick coat of hypervariable proteins called Variant Surface Glycoproteins (VSGs) of which there are over ~ 2000 (Muller et al. 2018). The vast majority are found in subtelomeric regions, with smaller numbers in specialised regions (called Expression Sites; ES), primed for VSG expression. ES VSGs are normally seen early during infection with subtelomeric VSGs detected later. In T. brucei, one ES is active meaning a single VSG is expressed on the surface of each cell. By stochastically changing which VSG is expressed in a process known as antigenic variation (Horn, 2014), the parasite can escape being recognised.
The underlying pathways which coordinate Trypanosome antigenic variation are complicated and there is still much to understand. However, DNA repair processes are needed, including homologous recombination (HR), a common pathway used to repair breaks across two DNA strands (a double strand break; DSB) by allowing similar or identical DNA sequences to be located and exchanged. The majority of VSG switching relies on HR to exchange one VSG for another. In other organisms, HR relies on a sensing complex known as MRN (MRE11-RAD50-NBS1) which can bind to DNA lesions and direct repair.
Given the importance of HR in Trypanosoma antigenic variation, how the MRN complex could act in this process was unknown. By investigating the roles of RAD50 and MRE11 in these parasites, Mehnert et al. reveal a role for both proteins in wide-spread and spontaneous DNA repair by promoting efficient HR and preventing repair occurring by alternative methods. Additionally, Mehnert et al. uncover a pathway which uses RAD50 activity to moderate HR activities during VSG exchange which could allow the parasite to maximise their vast collection of VSGs.
Key Findings:
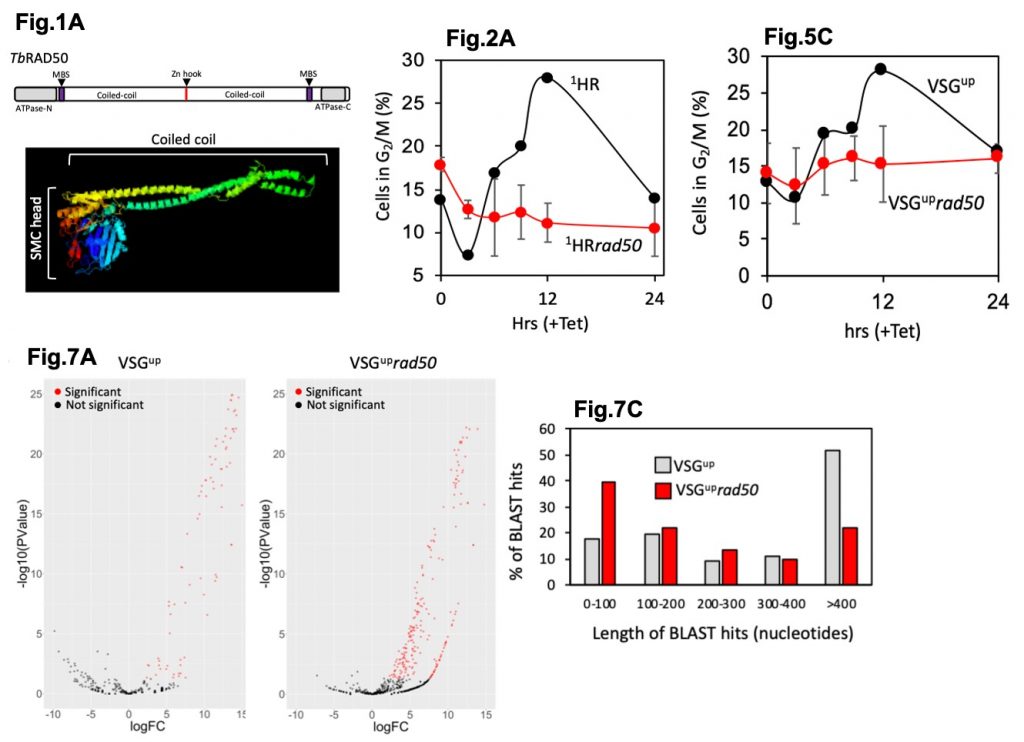
1) Cells lacking RAD50 or MRE11 grow poorly and largely fail to repair DSBs that occur within chromosomes. Mehnert and colleagues removed RAD50 or MRE11 from a cell line in which a DSB can be induced on a single chromosome with a tetracycline inducible meganuclease called I-SceI (Glover et al. 2008). In cells lacking RAD50 or MRE11, generating a DSB within a chromosome resulted in less than 3 % of cells surviving (compared with 48 % from the control line).
2) DNA damage signalling is impaired in cells lacking RAD50 or MRE11. In both mutants, reduced numbers of both phosphorylated H2A and RAD51 foci (markers of genotoxic stress and HR activity respectively) were observed after induction of an intra-chromosomal DSB suggesting these proteins are involved in the response to DSBs which may form spontaneously within chromosomes.
3) Without RAD50 or MRE11, most T. brucei parasites do not efficiently repair intra-chromosome DSBs by HR. Using a PCR based approach followed by sequencing to understand how the double strand break was repaired in surviving cells, Mehnert and colleagues reveal ~ 80 % of these parasites instead use a more error prone pathway requiring called microhomology mediated end joining (MMEJ). MMEJ only requires small regions of homologous sequence to fix broken DNA strands.
5) In contrast, if a double strand break is generated in close proximity to the actively expressed VSG gene, cells lacking RAD50 survived better than control cells despite reduced levels of H2A phosphorylation indicating their ability to signal a lesion may be compromised. Paradoxically, this data suggests that cells lacking RAD50 are likely more able to initial DNA repair in this region. In direct contrast, when the same experiment was performed in cells lacking MRE11, a worse survival rate was reported.
6) RAD50 can moderate access to subtelomeric VSGs by prioritising HR at a DSB near the expressed VSG. In cells lacking RAD50, repair mostly occurs by MMEJ using short regions of similarity but RNAseq analysis revealed a greater number of VSG coming from the subtelomeres. In control cells, > 400 bp regions were used to repair the DSB near the VSG. In cells lacking RAD50, 100 bp regions were instead used indicating shorter regions were used for repair. Shorter homology regions can be seen near subtelomeric VSGs and could explain the increase in subtelomeric VSG transcripts. This data suggests RAD50 limits access to the subtelomeric VSGs.
What I liked about this preprint:
I enjoyed reading this preprint by Mehnert and colleagues. African Trypanosome antigenic variation is a complex process and how the parasite balances necessary DNA repair activities with its VSG switching requirements is intriguing and often reveals how such pathways can be exploited for different purposes. Often the stochastic nature of this process leads to the misconception that this process is not controlled. However, by studying the MRN complex in T. brucei, the authors (in support of prior studies) demonstrate that molecular restraints operate to keep VSG switching in check.
Questions for the authors:
- Your data shows that loss of either MRE11 or RAD50 results defective growth and DNA damage signalling (Fig. 1-3) attributing this affect to the formation of spontaneous DNA breaks within chromosomes. However, deletion of neither is lethal. Firstly, would you expect a double null mutant cell line to be non-viable? Secondly, given MMEJ is an error prone pathway, could you comment on what you might expect to find, in terms of genome architecture changes, if you performed whole genome sequencing on cells?
- Following on from the last question, where do you think spontaneous DSBs are more likely to form in brucei within chromosome internal sites?
- When a DSB is generated in the ES of the active VSG, your data shows a divergence of function between MRE11 and RAD50. Given they generally operate as a complex, does your data suggest MRE11 and RAD50 can act together and in separate pathways in T. brucei? If so, does your data suggest the possibility of ‘two’ HR pathways? Or repurposing of the HR pathway in Tryps? Perhaps, one for driving VSG switching and one for chromosome internal breakages?
References:
- Muller et al. Genome organization and DNA accessibility control antigenic variation in trypanosomes. Nature (563):121-125 (2018)
- Horn, D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 195(2):123-129 (2014)
- Glover, L. et al. Sequence homology and microhomology dominate chromosomal double-strand break repair in African trypanosomes. Nuc. Acid. Res. 36(8): 2608-18 (2008)
doi: https://doi.org/10.1242/prelights.19019
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
preLists in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |