








FORK-seq: replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing
Posted on: 27 April 2020
Preprint posted on 10 April 2020
Article now published in Genome Biology at http://dx.doi.org/10.1186/s13059-020-02013-3
Following the FORKseq’s in the road. Paving the way for high-resolution mapping of DNA replication.
Selected by Jennifer Ann BlackCategories: genomics
Background:
To duplicate their genome on time, many Eukaryotes initiate DNA replication at multiple different sites on their chromosomes, known as origins of replication. When replication is initiated, the movement of the machinery required for replication forms structures known as a replication forks, which move bi-directionally from the origin. When they converge together, DNA replication can terminate (1). Most genome-wide approaches to study DNA replication are performed on cell populations. This means we lose resolution and important information on DNA replication events within individual cells. Here, Hennion and colleagues have developed a new protocol for examining single molecule DNA replication using the recently developed MinION nanopore sequencing technology from Oxford Nanopore Technologies (2). This technology can allow the sequencing of native single DNA strand in real time. When each DNA base enters and travels through the ‘nanopore’ channel in the flow cell, changes in the surrounding electrical field occur and corresponding electrical signature can be matched to a DNA base. Using the yeast Saccharomyces cerevisiae (S. cerevisiae) as a model, cells were labelled with bromodeoxyuridine (BrdU), an analogue of the DNA base thymidine (3), to track DNA replication on single strands of DNA by analysing location of BrdU incorporation and orientation of BrdU-abundance gradients. They term this approach ‘FORKseq’. Their data reveals a high-resolution and genome-wide picture of DNA replication events in yeast.
FORKseq takes a similar approach to the recently published D-NAscent method (4) which allows the examination of single molecule DNA replication events using BrdU incorporation and nanopore sequencing. The differences between the two techniques lie predominantly in their computational pipelines and their BrdU labelling strategies.
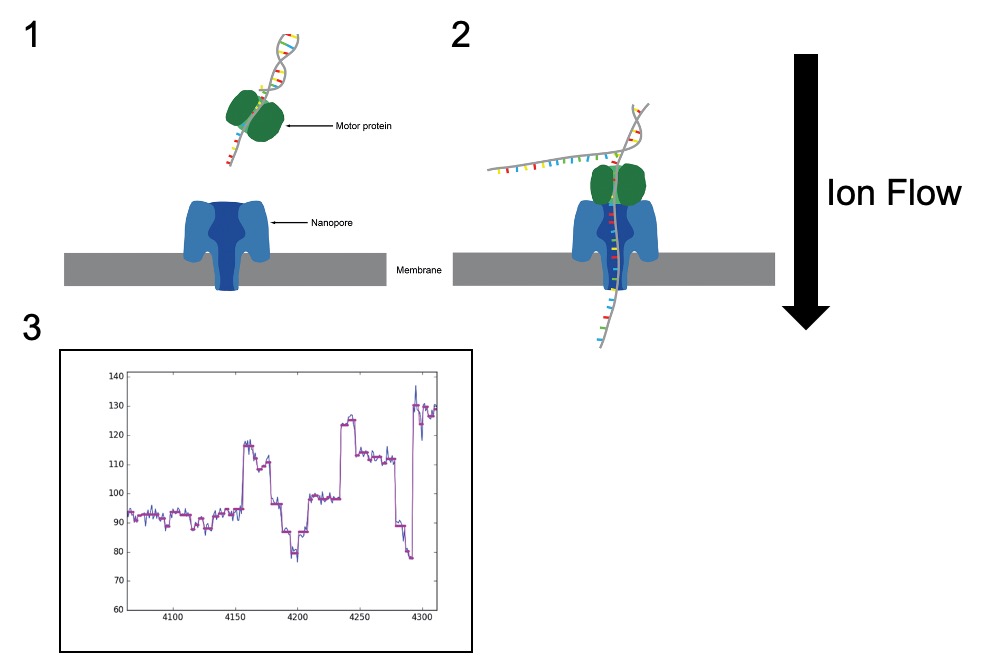
Figure 1. (1) The ‘Nanopore’ embedded in the membrane (blue). An ion current is flowed across the membrane. When the DNA library is applied to the membrane containing the Nanopore, the adaptors can bind to ‘tethers’ on the membrane surface. (2) A motor protein (green) helps to guide the DNA molecule through the pore. (c) As bases enter the Nanopore, the disrupted the electrical current can be read as patterns unique to each base allowing base calling to occur. Adapted from Leggett and Clark, 2017.
Key Findings
Nanopore sequencing technology can distinguishing between thymidine and its base analogue BrdU in a native single strand of DNA.
By sequencing primer extension products containing the presence or absence of BrdU incorporation, a difference between the electrical charge of BrdU and thymidine could be detected by the MinION sequencer using custom python scripts (Fig. 1). Next, by implementing two different machine learning algorithms (CNN and TM), the authors assessed abundance of BrdU incorporation within 100 bp windows, with their bioinformatics approach largely agreeing with average BrdU abundance detected by their mass spectrometry analysis confirming their analysis approach is viable.
FORKseq can determine replication fork direction
To look for replication fork progression, yeast cells were cultivated (pulsed) in BrdU and thymidine conditions for 2 mins then ‘chased’ using thymidine. When the resulting DNA was sequenced, the authors observed regions in which sharp transitions from low BrdU abundance to high BrdU abundance occurred (Fig.2).
Using their machine learning approach, they found these transitions revealed the direction of the replication fork, which they confirmed using a previously published approach known as OK-seq (4,5). They also show they can resolve these sites to within ~ 200 bps of the start and initiation site giving a high-resolution picture of replication events.
FORKseq can map individual replication initiation and termination sites in the yeast genome
Replication initiation and termination sites have been previously mapped throughout the yeast genome. Using FORKseq data, the authors largely confirm the positionings of these sites and additionally identified regions of initiation (9 %) and termination (18 %) that were likely missed previously due to a lack of sensitivity for sites of infrequent usage. Here, they reveal that these additional initiation sites are new sites of DNA replication initiation out with the known origin of replication supporting the model that yeast DNA replication can commence from canonical origins (91%) and non-canonical origin sites (9%). The additional termination sites were located within regions previously only associated with DNA replication initiation (Fig. 4-7).
FORKseq is adaptable for use in other Eukaryotic systems
The FORKseq approach relies on BrdU incorporation as a measure of DNA synthesis. BrdU has been successfully used widely in other Eukaryotic organisms to study DNA replication (3) and established published protocols are available for reference in the usage of this base analogue. Though the authors do acknowledge that organisms with larger genome could likely prove challenging to analyse due to restrictions on the throughput achieved by the MinION sequencer.
What I liked about this preprint:
DNA replication often studied in a population of cells as examining single molecules of DNA can be challenging meaning we are likely more familiar with a lower resolution representative of this process. I really enjoyed reading this article by Hennion and colleagues because of their approach to tackle this lack of resolution. They present their findings clearly, acknowledge their protocol in the context of the field and other techniques developed for similar purposes and employ clever strategies to maximise their datasets.
Questions for the authors:
- Your approach could be used to address how replication dynamics are affected by the presence of exogenous replication stress, such as the addition of chemicals like Hydroxyurea. Do you foresee any challenges or limitations for FORKseq in the analysis of such data?
- As mentioned, nanopore sequencing can be challenging to ensure reproducibility between experiments. What did you find most challenging about the sample preparation and/or the data analysis to minimise variation between experiments?
References:
(1) Fragkos, M., Ganier, O., Coulombe, P & Mechali, M. DNA replication origin activation in space and time. Nature Reviews Molecular Cell Biology, 16 (2015)
(2) https://nanoporetech.com/applications/dna-nanopore-sequencing
(3) Cavanagh, B.L., Walker, T., Norazit, A. & Meedeniva, A.C. Thymidine analogues for tracking DNA synthesis. Molecules, 16(9) (2011).
(4) Muller, C.A., Boemo, M.A., Spingardi, P., Kessler, B.M., Kriaucionis, S., Simpson, J.T. & Nieduszynski, C.A. Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nature Methods, 16 (2019).
(5) McGuffee, S.R., Smith, D.J. & Whitehouse, I. Quantitative, genome-wide analysis of Eukaryotic replication initiation and termination. Molecular Cell, 50(1) (2013).
(6) Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Sylvain M, Thermes C, Chen CL, Hyrien O. Replication landscape of the human genome. Nature Comm., 7, 10208 (2016).
Figure reference:
Leggett, R.M & Clark, M.D. A world of opportunities with nanopore sequencing. J Exp. Botany., 68, 20 (2017).
doi: https://doi.org/10.1242/prelights.19484
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |