








Modeling transcriptional profiles of gene perturbation with a deep neural network
Posted on: 13 February 2024 , updated on: 30 September 2024
Preprint posted on 16 July 2021
Can we predict DNA perturbations from RNA expression values? This exciting manuscript finds that training a deep learning model with 978 transcript levels is sufficient to predict which gene got silenced following shRNA knockdown!
Selected by Raquel MoyaCategories: bioinformatics, cancer biology
Context
The ability to predict genetic perturbation(s) from the gene expression profile of a cell line or tissue can provide valuable genomic information to researchers, clinicians, and patients, while avoiding expensive sequencing and time-consuming genomic analyses. Before such a prediction tool can be used in a clinical setting, however, researchers must extensively validate that it can correctly infer known genomic perturbations in existing datasets. Deep learning models have recently been developed and applied to the task of accurately predicting genomic data (e.g., Xpresso, Basenji, Enformer, and Borzoi).1–4 In line with this, the authors of this preprint have trained a deep learning model using the Connectivity Map (CMap) dataset, which is a large collection of transcriptomic profiles in response to many different compounds and genetic modifications. One goal of this deep learning model is to improve the conventional algorithm developed by CMap that determines a perturbation target gene by comparing its gene expression profile against other profiles using a similarity metric.
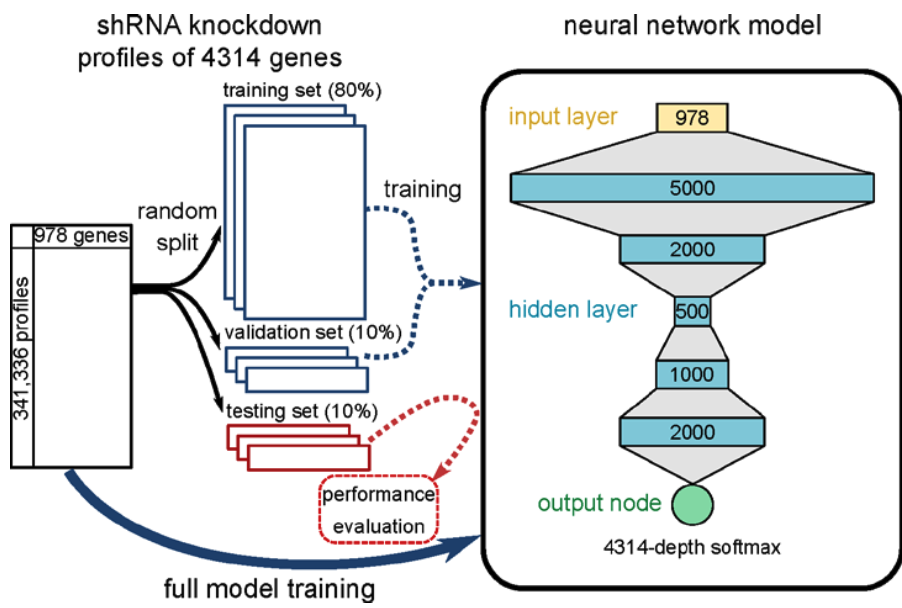
Methods
The dataset used in this study derives from short hairpin RNA (shRNA) knockdown experiments conducted in 9 cell lines (GEO accession GSE106127) conducted as part of the NIH Library of Integrated Network-Based Cellular Signatures (LINCS) initiative.5 Gene expression profiles for these experiments were generated using the L1000 platform, which is a multiplexed gene expression assay that involves measuring transcript levels with fluorescent beads using a flow-cytometry scanner. Each bead was analyzed both for its color (indicating gene identity) and fluorescence intensity of the phycoerythrin signal (denoting gene abundance).5 The authors developed a deep learning model with a convolutional input layer with 978 nodes (one for each gene), 5 hidden layers of different sizes, and one output node which outputs a vector of probabilities for each possible perturbation target gene (n = 4,313). The model was trained on 80% of the total 341,336 gene expression profiles, where 10% was reserved for model validation and 10% for testing. The aim of the model during classification was to accurately predict the perturbation target gene for each shRNA knockdown.
Key Insights
After training, the model performed with an average AUROC across all classes of 0.99 on the testing data. The authors applied this trained model to a similar, but inherently different, CRISPR knockdown dataset for which the model performed less well (average AUROC across all classes = 0.6078). Attempts to improve performance on the CRISPR knockdown dataset did not change its initial performance. Taken together, this manuscript has demonstrated a deep learning adaptation of a more conventional algorithm that performed well to predict a shRNA target gene. Generally, it is exciting to see the application of deep learning frameworks to gene expression data because the complexity of the transcriptome is potentially matched by the power and scalability of a large network.
Questions for the authors
This intriguing study raised some questions about its application and the generalizability of the presented modeling approach.
- Landmark genes:
- The authors could touch on how the original L1000 paper used the measurement of the ~1,000 “landmark” transcripts to infer the remainder of the transcriptome. Only the expression values of the 978 chosen “landmark” genes were involved in training the deep learning model in this manuscript, however it may contextualize the model’s performance to mention that inference of the remainder of the transcriptome using the “landmark” set was accurate for only 81% (n = 9,196/11,350) of inferred genes. Thus, 17% of genes can’t be inferred from the L1000 transcript set. This could affect model performance and make a certain perturbation target gene appear as a different one. A larger or different set of “landmark” transcripts that recapitulates the transcriptome better may facilitate improved model performance.
- Off-target effects: The original L1000 paper discusses the possibility of their platform being able to analyze off-target effects. They compared “similarity between shRNAs targeting the same gene (“shared gene”) and shRNAs targeting different genes but sharing the 2-8 nucleotide seed sequence known to contribute to off-target effects (“shared seed”)”.5 Their conclusion was that the shared gene similarity was only slightly greater than random. In contrast, shared seed pairs were dramatically more similar to a null distribution, indicating that the magnitude of off-target effects exceeds that of on-target effects for shRNA knockdown.
- It seems tricky to train a model to accurately predict the perturbation target gene given the amount of off-target effects captured by the training data. Could the model be adapted model to mitigate the influence of off-target effects? One idea could be to somehow combine or reduce the noise in the gene expression profiles from different shRNAs that target the same gene. This could improve model performance because two different shRNAs targeting the same gene might have similar on-target effects and different off-target ones.
- How much variability around a measurement exists in this dataset for different shRNAs targeting the same gene? How concordant are their profiles?
- Imbalanced classes:
- Genes with low accuracy had a small number of shRNAs. Could the authors control for the number of shRNAs per gene in the training data by filtering or down-sampling some target genes?
- How imbalanced are the classes? The authors could show the number of shRNAs per target gene and whether this is uniform across target genes, for example.
- The authors report that the average accuracy of the trained model on the test set is 74.9%. It would also be great to show AUPRC (area under the precision-recall curve) too. The authors report AUROC, which is great, however caution could be used because high recall can be achieved at a very low false positive rate owing to the large number of negatives in the test set, making it easy to obtain a high AUROC even when false positives vastly outnumber true positives (i.e., high false discovery rate).
- Generalizing to a CRISPR knockdown dataset:
- It is possible that a model trained for one context can often perform poorly on another. While the L1000 platform was also used to measure gene expression profiles in the CRISPR knockdown dataset, there are biochemical and experimental differences between shRNA and CRISPR knockdowns.
- Is it possible that the model has memorized the training data? Certain profiles in the training data could exhibit similar profile-perturbation relationships in the testing data, such as the same shRNA target gene. I wonder how partitioning entire biological groups into either the training or test set would affect results.
- The network is fully connected and such networks tend to memorize training data entirely when given enough time. Could the model architecture be contributing to memorization of the training data and lower generalizability?
- CGS query method: The CMap dataset can be used to infer a gene perturbation from a gene expression profile by entering a query profile that is compared to profiles in the dataset using a similarity metric. Does the deep learning model outperform this method? I’d be curious to know how the new model performs compared to the query method’s performance.
References
- Agarwal, V. & Shendure, J. Predicting mRNA Abundance Directly from Genomic Sequence Using Deep Convolutional Neural Networks. Cell Rep. 31, 107663 (2020).
- Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
- Kelley, D. R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 (2018).
- Linder, J., Srivastava, D., Yuan, H., Agarwal, V. & Kelley, D. R. Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation. 2023.08.30.555582 Preprint at https://doi.org/10.1101/2023.08.30.555582 (2023).
- Subramanian, A. et al. A Next Generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437-1452.e17 (2017).
- Weirauch, M. T. et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31, 126–134 (2013).
doi: https://doi.org/10.1242/prelights.36495
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the cancer biology category:
Nature-Inspired Nanoparticle Adiposomes Enable Targeted Delivery of Hydrophobic Drug for Anti-Cancer Treatment
Elizabeth Pyman
Targeting CXADR-mediated AKT signaling suppresses tumorigenesis and enhances chemotherapy efficacy in Ewing sarcoma
TheLangeLab et al.
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |