








Myc instructs and maintains pancreatic adenocarcinoma phenotype
Posted on: 8 March 2019
Preprint posted on 21 February 2019
Article now published in Cancer Discovery at http://dx.doi.org/10.1158/2159-8290.CD-19-0435
Categories: cancer biology
Background
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease with a 5-year survival of 5%. Poor prognosis is a result of the aggressive nature of the disease with as many as 90% of patients diagnosed with unresectable cancer that is resistant to chemotherapy. A key feature of PDAC is a dense fibroinflammatory stroma which can make up to 90% of the tumour bulk. This produces a complex microenvironment of mesenchymal, endothelial, inflammatory and immune cells with research beginning to understand the complexities of these interactions (see Tape et al., 2016).
It is currently thought that pancreatic ductal adenocarcinoma (PDAC) arises through a stepwise progression of precursor lesions called pancreatic intraepithelial neoplasias (PanINs). PanINs are characterised by the sequential accumulation of mutations. Initially, the oncogene KRAS is mutated and this is typically followed by inactivation of tumour suppressors such as TP53 or CDKN2A. Although we know the aberrations associated with PDAC progression, the specific function at different stages of the disease remains unclear.
Key findings of the preprint
Translocation of Myc to the nucleus drives rapid immune infiltration and disease progression.
The oncogene Myc is frequently activated in PDAC and has a plethora of downstream effects that culminate to mediate cell proliferation and drive PDAC progression (Hessman et al., 2015). To address the question of how Myc activation affects tumour progression the authors generated and characterised a new mouse model of pancreatic cancer. They combined the Pdx1-Cre; LSL-KrasG12D (KC) model with a homozygous Rosa26-LSL-MycERT2 transgene. This resulted in KrasG12D expression in the pancreas and a controllable MycERT2 variant of c-Myc which translocates to the nucleus, and is hence active, upon the injection of tamoxifen. This provided temporal control over Myc activation and novel insight into the role of Myc in the progression of PDAC.
Firstly, because the KC model develops precursor lesions in a defined timeframe, MycERT2 could be activated for 3 weeks when only early stage lesions are present in the pancreas. Interestingly, this induced a dramatic and rapid progression to PDAC with extensive stroma. Characterisation of the stroma found an avascular, hypoxic, desmoplastic stroma infiltrated with leukocytes. Strikingly, increased proliferation and changes to the stroma were observed after 1 day of Myc activation (Figure 1). As a control, MycERT2 activation alone in the pancreas only promoted a moderate increase in proliferation.
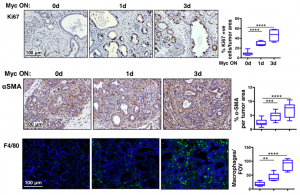
Figure 1: Myc activation drives rapid proliferation (Ki67), stellate cell activation (aSMA) and recruitment of macrophages (F4/80) to PanIN lesions.
Myc elicits multiple signals in PanIN’s to promote immune infiltration
As MycERT2 is only expressed in the tumour epithelium the authors hypothesised that the signals driving stromal changes arise from the epithelium. To better understand these signals, arrays were carried out 6 hours after tamoxifen administration to activate Myc in PanINs. They identified several signals originating from the epithelium associated with immune infiltrate: the macrophage chemoattractants CCL9 and CCL2 plus the neutrophil attractant CXCL5.
Upregulation of the AXL kinase ligand, GAS6, was also observed. Inhibition of GAS6 or AXL kinase inhibited Myc-induced neutrophil influx but did not affect CXCL5 which led the authors to suggest this is a novel, Myc-driven neutrophil recruitment pathway. Together, this analysis highlighted several candidate signals from the epithelium responsible for the accumulation of stroma.
Myc de-activation triggers disease regression and apoptosis
One advantage of the MycERT2 model is that Myc can be turned off. Following 3 weeks of activated Myc, which induced PDAC in the KC model, Myc was de-activated by stopping the administration of tamoxifen. Surprisingly, this led to regression of all adenocarcinomas. Next the study tested whether Myc dependency is a common feature of PDAC. For this the mutant KrasG12D and p53-deficient model of PDAC was combined with the Omomyc dominant negative Myc dimerization mutant. Importantly, blocking Myc by this method also triggered regression of PDAC tumours.
To assess the earliest changes following de-activation of MycERT2, the authors looked at PDAC tumours 1 and 3 days following the removal of tamoxifen. They observed rapid elimination of macrophages and neutrophils, loss of desmoplasia and decreased hypoxia. In addition to this massive stromal reversion the tumour epithelium exhibited decreased proliferation but also increased apoptosis (Figure 2).
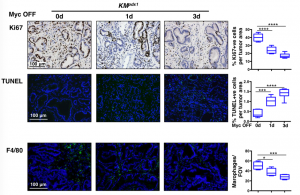
Figure 2: Analysis of PDAC following de-activation of Myc caused a significant decrease in proliferation (Ki67) and macrophages (F4/80). Moreover, increased apoptosis (TUNEL staining) was observed in the tumour epithelium.
What I like about this preprint
Lots of animal models of pancreatic cancer activate oncogenes and delete tumour suppressors simultaneously and examine tumourigenesis. But PDAC appears to initiate with KrasG12D mutations and then accumulates further aberrations. The novel control of Myc by fusion to the ERT2 domain provides very nice temporal control and sequential activation of Myc months after Kras activation.
The other benefit of this approach is you can de-activate Myc and look at both the acute and longer term effects of this withdrawal. This demonstrated significant changes to the tumour and stroma in only 24 hours!
Finally, it’s really interesting that simply switching on or off Myc had this large, orchestrated effect leading to tumour progression or regression, which the authors propose could be part of some coordinated physiological process.
Questions to the authors
If Myc hacks the endogenous regenerative program what do you think would happen if you combined your Myc model with other models of regeneration such as caerulein induced pancreatitis?
Why do you think removal of Myc leads to apoptosis of epithelia in vivo?
Did you see an effect on metastasis upon induction of Myc?
I think this idea of Myc hacking into a physiological injury resolution program is really exciting, do you think it would be possible to pharmacologically promote this program?
References:
Hessmann, E., Schneider, G., Ellenrieder, V., & Siveke, J. T. (2016). MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene, 35(13), 1609.
Tape, C. J., Ling, S., Dimitriadi, M., McMahon, K. M., Worboys, J. D., Leong, H. S., … & Jørgensen, C. (2016). Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell, 165(4), 910-920.
doi: https://doi.org/10.1242/prelights.9267
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the cancer biology category:
Targeting CXADR-mediated AKT signaling suppresses tumorigenesis and enhances chemotherapy efficacy in Ewing sarcoma
TheLangeLab et al.
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
preLists in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |