








Self-reporting transposons enable simultaneous readout of gene expression and transcription factor binding in single cells
Posted on: 20 February 2019 , updated on: 25 July 2020
Preprint posted on 1 February 2019
Article now published in Cell at https://www.cell.com/cell/fulltext/S0092-8674(20)30814-X
Making a mark on gene regulatory networks: A method using transposon insertions directed by a transcription factor allows simultaneous mapping of transcription factor binding sites and gene expression in single cells.
Selected by James BriscoeCategories: genomics
Background
One of the aspects of preprints that I’ve found particularly useful is the rapid communication of innovative new methods. This is particularly true in fast moving fields such as single cell assays. I think the work by Moudgil et al is a good example of this.
At the heart of developmental mechanisms are gene regulatory networks – collections of transcriptional regulators that interact with each other, through the cis-regulatory elements they bind, to control gene expression and hence cell identity and function. Developing methods that allow the simultaneous assay, in individual cells, of the transcriptome and the genomic binding pattern of specific transcription factors (TF) would offer new insight into gene regulatory networks. In this preprint, Moudgil et al develop a method to do just this.
The method
To identify TF binding sites the authors previously described a technique that is based on a fusion between a TF of interest and a transposase [1]. The TF-transposase chimera is introduced into cells along with a reporter-harbouring transposon. As a result, the TF-transposase targets deposition of the reporter-transposon to DNA near the TF binding sites. The authors refer to these insertions as “calling cards” that can be amplified from chromatin and the locations determined by high-throughput sequencing of genomic DNA.
To make the technique compatible with transcriptome assays, the authors extended the technique by developing “self-reporting transposons (SRTs)”. To do this they removed the polyadenylation signal from the reporter-transposon and added a ribozyme after the terminal repeat to minimize reads from the non-integrated reporter. These clever tricks allow transcription of the reporter gene through the transposon into the flanking genomic sequence. The location of an insertion event can then be identified from mRNA by the sequence of the 3’ untranslated regions (UTRs) of reporter gene transcripts.
The authors first validate the technique in populations of cells transfected with the transcription factor SP1 fused to the transposase by demonstrating that calling cards sequenced from mRNA UTRs overlap with positions in the genome that are known to bind SP1. The transposase used, piggyBac, naturally interacts with the bromodomain protein BRD4, which itself associates with acetylated histones and active enhancers. The authors turn this potential bug into a feature by demonstrating that, if not fused to a specific TF, piggyBac can be used to map BRD4 bound regions in the genome using the SRT technique.
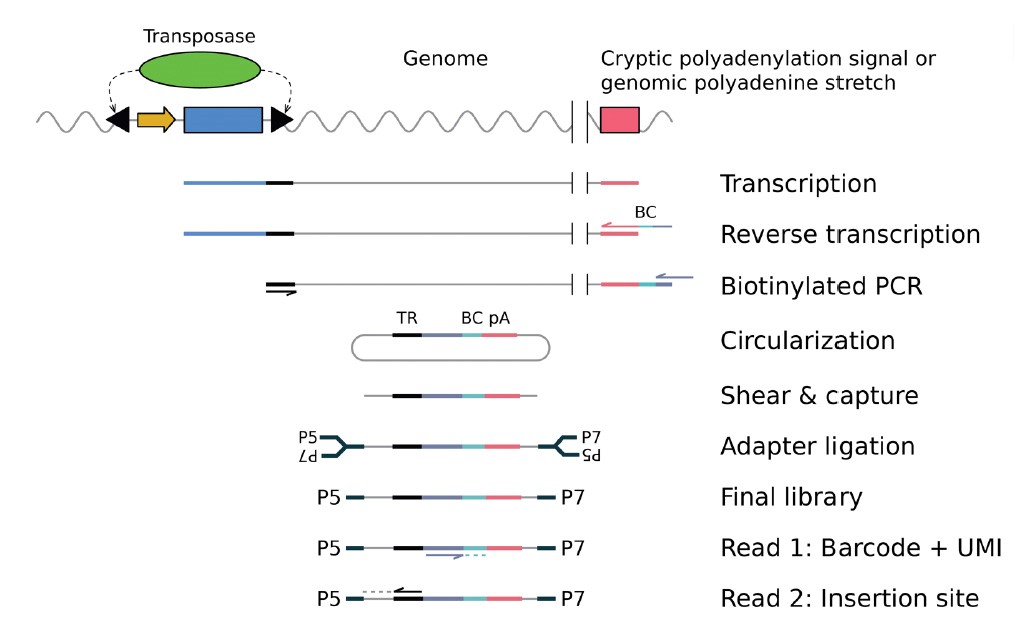
Finally, the authors modify their protocols to apply it to single cells – scCC (single cell Calling Cards). mRNA from single cells transfected with the SRT system was used to recover both the call cards, indicating the locations of transposon insertions, and the transcriptome. The cell barcodes from the single cell library preparation allow transcriptome data to be paired with call cards and hence transposon insertions assigned to specific cell types. Proof of principle experiments in cell lines were followed by mapping of Brd4 binding and gene expression in individual cells from the mouse cortex. They demonstrate differential Brd4 binding in excitatory neurons located in different layers of the cortex, providing evidence that SRT can be used to map transcriptional regulators in situ.
Why I like the preprint
I think the approach is elegant and original with the potential for further refinement. It has similarities with some other recently developed techniques, such as targeted DamID (e.g.[2]), which identifies TF binding sites using a TF fused to DNA methyltransferase that methylates GATCs in the neighbourhood of binding. But scCC allows the simultaneous recovering of mRNA as well as TF binding location in individual cells, this has the potential to infer the link between TF binding and gene regulation. Moreover, the approach is of broad interest as it is sufficiently flexible to apply to almost any TF, in any cell type or tissue, in any species.
Many modifications to the system can be imagined. The authors mention the idea of using calling card insertions as molecular records of cell lineage or specific cellular events. Another possibility would be to use two or more distinguishable reporter-transposons, introduced at different times, to examine temporal changes in binding. It’s also possible to imagine using orthogonal transposase-transposon pairs to simultaneously monitor the binding of two TFs in the same cell.
Questions and open issues
As the authors point out, potential limitations are the sparsity of the data from single cells and the inherent bias in the insertion preferences of the transposase. In this context, it would be interesting to know if enhancers that produce eRNAs (~25% of enhancers) might be captured more frequently. Tweaks to the system and scaling up the datasets could address some of the shortcomings. Ultimately, analyzing the correlation between TF binding and the activity of individual genes in populations of many single cells would offer fantastically rich datasets from which to make gene regulatory predictions.
The current system relies on the ectopic expression of the TF-transposase fusion protein. Whether this results in aberrant binding of the TF to sites not normally occupied or whether the expression of the TF-transposase fusion protein has dominant effects that alter the state of the cells will depend on the details of the TF and cell types. Developing the system to allow the regulated expression from an endogenous gene would be one way around this limitation.
In the study, the authors use the interaction between PiggyBac and Brd4 to their advantage. However, this could also be a limitation as it might result in a high background or confounding results that complicate the identification of binding events that are specific to a TF of interest. It was unclear how much the Sp1-PiggBac fusion protein is recruited to Brd4 bound sites. Modifications that reduce or abrogate the Brd4 interaction, perhaps by using alternative transposases, would eliminate this concern. In addition, I’d be interested in seeing a comparison between BRD4 binding sites identified in the scCC system and other techniques, such as scATACseq, that mark accessible chromatin, to see how much overlap there is.
References
- Wang H, Mayhew D, Chen X, Johnston M, Mitra RD. (2012) “Calling cards” for DNA-binding proteins in mammalian cells. Genetics. 190:941-9
- Cheetham SW, Gruhn WH, van den Ameele J, Krautz R, Southall TD, Kobayashi T, Surani MA, Brand AH. (2018) Targeted DamID reveals differential binding of mammalian pluripotency factors. Development 145:dev170209
doi: https://doi.org/10.1242/prelights.8742
Read preprint (1 votes)
(1 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |