








The mechanism of artemisinin resistance of Plasmodium falciparum malaria parasites originates in their initial transcriptional response
Posted on: 23 June 2021
Preprint posted on 17 May 2021
Transcriptional profiles of artemisinin-resistant malaria parasite Plasmodium falciparum from endemic areas of Asia
Selected by Victoria AlonsoCategories: systems biology
Background
The parasite Plasmodium falciparum transmits malaria through the bite of Anopheles mosquitoes. Of all Plasmodium species, P. falciparum is the major cause of malaria. In 2019, it was responsible for 99.7% of estimated malaria cases in Africa, 50% of cases in South-East Asia Region, 71% of cases in the Eastern Mediterranean and 65% in the Western Pacific . The same year there were estimated 229 million cases of Malaria and 409.000 deaths worldwide.
Current antimalarial control is highly dependent on artemisinin combination therapies. Thus, it is very concerning that parasites with a decreased sensitivity to this drug have emerged, resulting in the delayed clearance of parasites from patients. This slowed parasite clearance is predominantly associated with nonsynonymous mutations in the P. falciparum kelch13 gene (PfK13). This lineage of artemisinin resistance is spreading throughout Asia and Africa, threating the efforts to treat and eradicate malaria. It is thought that artemisinin resistance involves multiple cellular and metabolic processes. Although many of these processes have been described in vitro, what´s going on in natural infections is less certain. In this preprint, Zhu and coworkers from different parts of the world (Thailand, Singapore, UK, Netherlands, Bangladesh, US, Myanmar, Laos, Vietnam and Israel) aimed to tackle this issue using a transcription-wide associated analysis. Transcription-wide associated analysis estimates the effect of each genetic variant on the activity level (or expression) of genes related to a disease. Thus, using this method allowed them to identify causative genetic variations of artemisinin resistance using parasites isolated from natural infections that occurred between 2016 and 2018.
Key findings of the preprint
Transcriptomic sequencing of the P. falciparum population in the Great Mekong Subregion (GMS, the region in which antimalarial drug resistance was first identified)
First, the authors isolated P. falciparum RNA from 577 patients’ blood samples without artemisinin treatment (zero-hour) and conducted DNA microarray and RNA-seq. For 459 patients, samples were also collected 6 hours after taking artemisinin. From the zero-hour samples, they were able to identify the distribution of the different life cycle stages of P. falciparum and found out that the ring stage (young intra-erythrocytic asexual stage) was predominant. The principal component analysis (PCA) of the zero-hour transcriptomic profile showed that the hours post infection variable generated the major differences across samples. Another interesting result obtained from the PCA is the separation of two major parasite groups according to geographic regions (eastern and western GMS). This division correlates with a specific lineage with the artemisinin-resistant C580Y mutation of PfK13 found exclusively in the eastern region (eGMS).
Transcriptome-wide associating study of artemisinin resistance
Next, the authors performed a transcriptome-wide associated analysis and identified specific genes, most likely acting together with PfK13 mutations, whose expression/activity is participating in the parasite resistance to artemisinin. They related the expression of each gene with the parasite clearance half-life and hours post infection. They identified 69 upregulated and 87 downregulated transcripts in the zero-hour samples. These transcripts correspond to genes whose biological function has been previously linked to artemisinin resistance, including protein and redox metabolism, functions linked to the digestive vacuole and mitochondria, and host cell remodeling. They collectively termed these genes as Artemisinin Resistance-associated Transcriptional Profile (ARTP).
Utility of ARTP as a marker of spread and evolution
To investigate the use of the ARTP as a representation of artemisinin resistance in eGMS, the authors performed a clustering analysis in the resistant parasite samples (identified by their slow clearance phenotype) and identified six clusters with a distinct geographical distribution. No bias was observed regarding lineage in any cluster. This suggests that the ARTP can be used as a fingerprint to investigate resistance to artemisinin.
In vivo transcriptional response of P.falciparum to artemisinin
To assess the transcriptomic response to artemisinin in vivo, the authors evaluated and compared the six-hour samples to the zero-hour samples. They identified 20 induced genes and 73 repressed genes in resistant parasites after 6 hours of treatment and 33 induced and 34 repressed genes in susceptible parasites with almost no overlap, suggesting a distinct transcriptional response to artemisinin. Finally, the authors compared their results with in vitro transcriptional responses reported in the literature, as summarized in Figure 1. This figure shows the functional assignment of resistance markers in different processes inside the parasite.
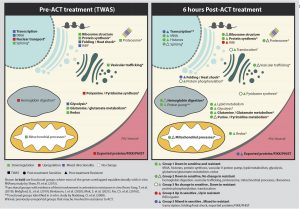
Figure 1: Functional assignments of transcriptional artemisinin resistance markers derived from TWAS analysis at zero-hour and that of genes repressed/induced after 6 hours of treatment with artemisinin. Colours represent transcriptomics directionality for each group and also the association to previous independent in vitro studies.
Summary
The data presented and analyzed by the authors suggest that there is a characteristic transcriptional pattern emerging in artemisinin-resistant P. falciparum parasites in Asia. They describe that the processes involved in artemisinin resistance are very complex and occur early in the asexual cycle of P. falciparum, diminishing the killing rate of the drug at an early stage.
What I liked about this preprint:
Malaria is a parasitic disease that is especially endemic in low-income populations, with a large health and economic impact on both the developing and developed world, causing millions of deaths every year. Resistance to antimalarial medicines is a recurring problem around the world. I think it is important to address this issue, because these diseases are often overlooked. Using high-throughput sequencing technologies, the authors of the preprint were able to identify specific genes involved in artemisinin resistance in P. falciparum. This could potentially be a first step in identifying a marker for evolution and spreading of drug resistance in malaria.
Future directions and questions for the authors:
Currently, many countries with a high burden of malaria have weak surveillance systems and are not in a position to assess disease distribution and trends, making it difficult to optimize general responses and respond to acute outbreaks. The authors suggest that it is possible to use this technology to perform Epidemiological Surveillance of artemisinin resistant parasites.
- How do you imagine it could be implemented? Particularly in low-income countries.
- Combination therapy is one of the best approaches that is currently being used to treat malaria. Have you considered analyzing resistance to the combination of drugs used for treatment using transcriptome-wide associated analysis?
- Why did you choose to compare the zero-hour transcriptome with the six-hour transcriptome? Could it be useful to take samples at earlier or later time points post treatment?
References
https://www.who.int/news-room/fact-sheets/detail/malaria
Cao C, Ding B, Li Q, Kwok D, Wu J, Long Q (2021) Power analysis of transcriptome-wide association study: Implications for practical protocol choice. PLoS Genet 17(2): e1009405. doi:10.1371/journal.pgen.1009405
doi: https://doi.org/10.1242/prelights.29731
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the systems biology category:
BAF complexes maintain accessibility at stimulus-responsive chromatin and are required for transcriptional stimulus responses
Dina Kabbara
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
preLists in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |