








High-throughput discovery and characterization of human transcriptional effectors
Posted on: 19 October 2020 , updated on: 23 October 2020
Preprint posted on 10 September 2020
Article now published in Cell at http://dx.doi.org/10.1016/j.cell.2020.11.024
Categories: systems biology
Background
Transcription factors (TFs) can generally be separated into two functional components – the DNA binding domain (DBD) and the effector domain. As its name implies, the DBD recognises and binds to its target sites in the genome, while the effector domain recruits cofactors to either promote or repress transcription. While DBDs are generally well-characterised and conserved, much less is known about effector domains. Effector domains tend not to be well-conserved and we know very little about their sequence features, making it difficult to predict which parts of the proteins are effectors and what cofactors they interact with. This in turn makes it difficult to understand how variants in these proteins cause disease. In this preprint, the authors set out to improve our understanding of repressor domains by developing a high-throughput assay that allows them to measure the activity of tens of thousands of repressor domains in human cells. Using this method, they screened for repressor domains in annotated and unannotated proteins as well as performed a deep mutational scan (DMS) of the KRAB repressor domain to find the residues important for its function.
Key findings
The authors first developed a high-throughput assay for repressor domain activity called HT-recruit. There are two key components to the assay (Fig 1). First, they generated K562 cells containing 9x TetO binding sites upstream of a strong promoter driving the expression of a reporter gene (which includes a synthetic surface marker). Then, a library of putative effector domains is synthesised and cloned as fusion proteins with rTetR in a lentiviral backbone, and the lentivirus pool is used to infect K562 reporter cells. The fusion protein library is recruited to the TetO binding sites upon Doxycycline treatment, where repressor domains will silence the reporter gene. Thus, cells containing repressor domains will no longer express the surface marker (OFF pool) and can be separated from cells that still express it (ON pool) using magnetic separation. The two pools of cells can then be sequenced, and the repressor activity of each domain is calculated as the ratio of domains in the OFF:ON pool.
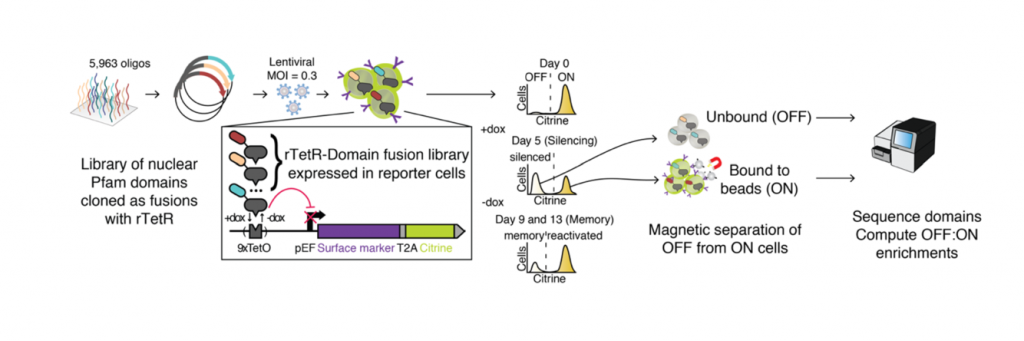
Fig 1: HT-recruit method. Adapted from Figure 1A.
For the first screen, the authors designed a library of candidate repressor domains using Pfam-annotated domains in human proteins that localise to the nucleus. They selected domains that were less than or equal to 80 AA, extending the domains with adjacent residues from native protein sequences for those less than 80 AA. They also added 861 negative controls – either random sequence or tiles of the DMD protein (not a nuclear protein). After removing domains that were poorly expressed, they identified 446 repressor domains at day 5. These domains represent 63 domain families and come from 451 proteins. Validation of the individual hits correlated well with the high-throughput measurements, demonstrating that HT-recruit can identify and quantify repressor activity accurately.
Using these measurements, the authors also managed to assign repressor function to some Domains of Unknown Functions (DUFs), which comprised about 22% of the Pfam-labelled domain families, expanding our understanding of proteins with unknown functions. Furthermore, they identified one random 80AA sequence (part of the negative controls) that had strong repressor activity.
Next, the authors looked deeper into KRAB domains, which is the largest family of TFs and includes some of the strongest known repressor domains. Most of the human KRAB domains were repressor hits, and repressors tended to interact with KAP1, a known co-repressor of KRAB. The repressors were also generally found in genes with a younger evolutionary age. To further understand KRAB domain function, the authors generated a deep mutational scan (DMS) library of the KRAB domain from ZNF10, which is the domain fused to Cas9 for CRISPR interference and thus widely used. Several single substitutions in the A-box were found to dramatically reduce repressor activity. Mutations in the B-box, however, show that the B-box partially contributes to KRAB silencing speed. Finally, mutations in the N-terminus leads to higher stability of the protein (and therefore higher expression), leading to seemingly increased repressor activity.
The second largest family of repressor domains identified in the screen was the homeodomain family. The authors looked more closely at the HOXL subclass which contains the Hox master regulators. They found that genes towards the 5’ ends of Hox clusters are stronger repressors and that the number of positively charged amino acids correlates with repression strength, suggesting that charge could play a role in regulating the repression domains in Hox genes.
The authors then modified HT-recruit to discover activation domains by changing the promoter of the reporter gene to the weak minimal CMV promoter. Using the same nuclear domain library, they found 48 hits from 26 domain families. Surprisingly, there were 4 hits in the KRAB domain family, including the strongest activator hit. Further functional analysis of individual domains shows that KRAB proteins are functionally diverse and can act as both transcriptional activators and repressors.
Finally, the authors used HT-recruit to find repressors in unannotated regions of proteins. They designed a library consisting of 80AA tiles of 238 proteins from silencer complexes and found repressor domains in 141 proteins, including known repressor domains as well as novel domains proteins with no annotated effector domains, showing that HT-recruit can successfully find and annotate effector domains in proteins.
What I liked
This is a massive piece of work that is rigorously conducted and provides an incredible resource for the community. The authors managed to obtain high-throughput, good quality data by generating a synthetic cell surface marker gene that enables efficient separation of ON and OFF cells. This preprint also addresses the gap in the literature by creating a list of repressor and activator domains, which will allow others to further curate and functionally dissect effector domains. I also particularly liked the fact that KRAB domains function as both activators and repressors, which challenges the assumption that domains with similar sequences will have similar functions. This also demonstrates the importance of categorising domains and proteins by their functions rather than by their sequence similarities.
Future directions and questions
The authors found that the RKKR motif and net positive charge was associated with repression strength in Hox domains but not in their full domain library. I am curious the authors have tried looking for enrichment of sequence features in repressor vs non-repressor domains. It seems unlikely that there would be one specific sequence feature, but perhaps there are classes of sequences that might look more similar.
Have the authors looked at the function of their tested domains in the context of the full-length protein? This is especially interesting for ZFP28 (which contains one repressor and one activator KRAB domain), is one dominant over the other in the full-length protein?
The measurements are quantitative and reproducible, suggesting that the position of the integrations do not really influence the output. Do the authors know how many integrations on average each library member has in order to neutralise position effect?
doi: https://doi.org/10.1242/prelights.25285
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the systems biology category:
BAF complexes maintain accessibility at stimulus-responsive chromatin and are required for transcriptional stimulus responses
Dina Kabbara
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
preLists in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |