








Large-scale analyses of human microbiomes reveal thousands of small, novel genes and their predicted functions
Posted on: 23 January 2019 , updated on: 24 January 2019
Preprint posted on 13 December 2018
Article now published in Cell at http://dx.doi.org/10.1016/j.cell.2019.07.016
The power of a computational magnifying lens – peering into diversity of proteins, large and small, encoded by microbes in our bodies reveals many novel small ORFs, their putative functions and hints on their evolution and diversity
Selected by Ganesh KadamurCategories: bioinformatics, microbiology, systems biology
Context: The coding region of genomes, commonly called Open Reading Frames (ORFs), have long been defined using a seemingly arbitrary minimum length cutoff (typically 50aa). Challenging this paradigm, studies over the past decade have shown widespread translation of stable products that fall below this threshold in both prokaryotes and eukaryotes, with functions in quorum sensing, development and calcium signaling, just to name a few. These proteins are encoded not only in intergenic regions, but also within annotated ORFs and have been named small ORFs (sORF). However, these studies have mostly relied on investigating well studied model organisms and so, extensive, large scale studies are notably missing.
Methodology: In this preprint, Sberro et al. tackle the question using large publicly available datasets and computational approaches. They mine metagenomes isolated from >250 human subjects as part of the Human Microbiome Project (NIH HMP I-II), running them through to a battery of analyses and computational pipelines. Using MetaProdigal, a prediction tool optimized to look for sORFs, they identify >2.5 million sORFs. Combining sequence based and domain analysis, these were grouped into ~400,000 clusters. To benchmark their methods, they search this set for ~30 sORFs from model organisms that have been well characterized and surprisingly find that almost half of these are absent in the human microbiome. To prune this list of ~400k clusters and increase confidence in their results, the authors utilize RNAcode, a program that incorporates evolutionary and mutational signatures amongst homologs to narrow down to bona fide sORFs. The authors then proceed to functional prediction and categorization of these sORF families using information about taxonomic specialization, intracellular location prediction based on sequence analysis and comparison to other environmentally sampled metagenomes. Also, as prokaryotic genes are commonly found in operons where functionally related genes are commonly clustered together, the genomic neighbourhood of sORF families was also analyzed for functional annotation. Together, this computational tour de force analysis has unearthed many novel sORF families, indicated putative functions and generates a vast body of hypotheses that can now be experimentally tested.
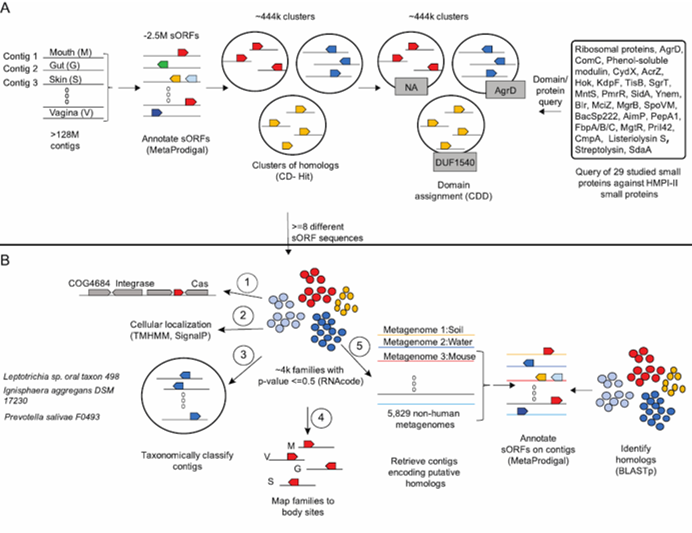
Pipeline for identification and prediction of function of sORFs from human microbiome metagenomic data (taken from Sberro et al., bioRxiv, 2018)
Key Findings:
- The human microbiome has >4000 sORF families of which almost 50% are not detected in species from other sequenced microbiomes (soil, water, mouse etc), thus highlighting the uniqueness of the microbiota that call our bodies home. In the process, the authors show that ~2400 families identified here are present in genomes included in the RefSeq database. However, more than 1000 such families had remained unannotated because of the arbitrary 50aa length cutoff.
- Some sORF families are more conserved than others. About 20 families are present in >50 species, whereas ~3000 families are found only in 10 or less species, suggesting that rapid evolutionary mutation and specialization is widespread amongst sORFs.
- A mere 4% of all identified sORF families possess annotated domains, underscoring the breadth of unexplored sequence and structure space amongst sORFs.
- 13 novel families are highly conserved across microbiomes isolated from different human niches (gut, mouth, skin) and thus likely encode essential housekeeping proteins. Almost half of these families are ribosome associated proteins – homologs of these families are also present in non-human microbiome species, supporting the prediction of them playing a critical role across phyla.
- No single protein family is present across all human niches sampled, implying niche-specific evolution of protein sequences and families. It is pertinent to note here that undersampling, of donor samples per niche, might bias this interpretation.
- About a third of novel sORFs are predicted to generate transmembrane and/or secreted proteins. Analysis of their genomic context suggests roles in quorum sensing, toxin-antitoxin systems and inter-cellular communication
- Clues from genomic neighbourhood identifies ~200 families as potential phage defense genes with roles in CRISPR response, and ~600 families that might mediate horizontal gene transfer events.
Why I like the work: This work extends previous findings that show the widespread yet unappreciated translation of small proteins, defined as <50aa in length. While earlier work was focused on a small number of well studied model organisms, this expands our knowledge of sORF families by orders of magnitude, uncovering uncharacterized protein domains (and thus folds) in the process. This work has generated a rich resource ripe for future exploration, with promise of discovery of new antibiotics, tools that could be developed to interrogate a plethora of cellular processes and possibly also innovative ways to design cell permeable proteins for drug delivery. The section where the authors clearly spell out potential pitfalls of the methods and conclusions of their work is also particularly commendable. Finally, this work is a great showcase of how combining different computational tools and pipelines can yield important insights into novel biology.
Future directions:
- Mass spectrometry to validate expression of predicted sORFs at protein level. This could also be complemented by techniques such as ribosome profiling.
- Re-analysis of the data to recognize co-occuring species, and presence of sORFs in both species with predicted quorum sensing roles in both – for example, a secreted sORF in one that acts as a signal transducer, and a signal receiving receptor, typically not a sORF, in the other species. This could enlighten on communication pathways employed by these species in specific niches to regulate inter-cellular crosstalk.
- Genetic studies in a wide range of species to test function. Development of a high throughput platform to study some families could be especially useful, for example sORFs predicted to play roles in quorum sensing. This would be contingent on ability to culture the species outside the body, and development of tools for genetic manipulation.
- Explore diversity and specialization of sORF families across individuals from different ethnic backgrounds, as research increasingly shows microbiomes widely vary based on diet, environmental factors etc. Such analyses could particularly help identify rapidly evolving sequences that are most important for local adaptation.
doi: https://doi.org/10.1242/prelights.7731
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
Also in the systems biology category:
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |