








Lightning Fast and Highly Sensitive Full-Length Single-cell sequencing using FLASH-Seq
Posted on: 12 August 2021
Preprint posted on 14 July 2021
Article now published in Nature Biotechnology at http://dx.doi.org/10.1038/s41587-022-01312-3
Categories: genomics
Background:
Understanding how differences in the DNA blueprint (genome) of a cell translate into differences in physical features (phenotype) can help us understand complex diseases like cancer (1). By investigating changes in gene expression levels i.e the ‘transcriptome’ of a cell, we can examine these effects. However, most transcriptomes examine populations of cells giving an overall picture of more common events. This means you miss information about what happens in individual cells, which is important, as individuals can have very different transcriptomes (2). It is possible to study the transcriptome of single cells using single-cell RNA sequencing (scRNA-seq), however most protocols are very time consuming, very expensive and, in some cases, lack sensitivity. Here, the authors modify a previously established scRNA-seq sample processing pipeline to create a protocol which is overall cheaper, more sensitive, and less time-consuming. Their approach ‘FLASH-seq’ will help improve the accessibility of scRNA-seq.
How does SMART-seq work?
FLASH-seq is based upon SMART-seq (switching mechanism at the 5’ end of the RNA template; 3). RNA is extracted from cells and converted into complementary DNA (cDNA) using a template switching reverse transcriptase (RT) enzyme. This enzyme adds a small sequence of bases (for example C nucleotides) to the 5’end of a full-length molecule of cDNA. It then stops and move to another molecule. This process is repeated until in theory all cDNA molecules now also have this small extra sequence of DNA at their 5’ end. These extra sequences, for example several C DNA bases, can now be recognised by another primer, allowing a second strand of DNA to be produced. Polymerase Chain Reaction (PCR) is then used to amplify the cDNAs. The cDNAs are then fragmented (‘broken up’) and prepared for sequencing using a next generation sequencing method. During this process an enzyme known as a transposase (tn5 transposase) adds a known short DNA sequence (Sequence Adaptor) required for the sequencing process. In a new protocol, it is now possible to also add Unique Molecular Identifies (UMIs) to cDNA molecules to enable researchers to find PCR duplicates (i.e duplicates from the same cDNA which enter different beads or flow cells during sequencing), count transcripts and help find rare variations of transcripts as some of these can be excluded during processing.
How does FLASH-seq improve the process?
Here, the authors develop their FLASH-seq protocol using human peripheral blood mononuclear cells (hPBMCs) from healthy individuals and from cultured HEK293T cells (an established line of human embryonic kidney cells). They sorted single cells using a flow cytometry approach into 96 well or 384 well plates. They compare FLASH-seq to established SMART-seq protocols.
They introduce several key modifications:
- Instead of using two steps to amplify the cDNA, they use one step to transcribe the RNA directly to cDNA (RT-PCR)
- They optimised the RT-PCR cycles
- They use an improved RT enzyme i.e less time is needed to make cDNA molecules
- They add extra nucleotides to the reaction to improve the number of cDNA molecules which have the extra 5′ sequences added by the template switching RT enzyme.
- Modified the primers used to copy the second strand to limit strand invasion. This is necessary as strand invasion results in shorter cDNA molecules as the RT process does not complete. This mean you would not get a complete cDNA molecule (4).
- No additional PCR amplification as the amplification during the RT-PCR reaction was adequate
- Modifications to the clean-up reaction after the RT PCR by limiting sample washing
These modifications improve:
Sensitivity
-
- 8x more cDNA than other SMART-seq approaches
- Full coverage of sequence across gene bodies
- Works in a challenging cell line like hPBMCs
Cost
-
- Around $1 per cell
- Uses smaller volumes of reagents which cuts the costs
- FLASH-seq outperformed other SMART-seq methods at these volumes
Adaptability
-
- It is possible to also add UMI’s to cDNA molecules during the FLASH-seq protocol
- Adding additional space sequences in the primers used to amplify the second strand and add the UMI meant they found less artifactual strand invasion events
- Can be run in a 96 well or 384 well plate format
- Can be handled by most automated lab liquid handling robots
Speed
-
- Sequence ready libraries in about 4.5 hours
- A more ‘rapid’ version of FLASH-seq known as FLASH-seq LA (low amplification), is also available. This approach allows minimal RT-PCR cycles and very limited purification of PCR samples but still achieves high quality results.
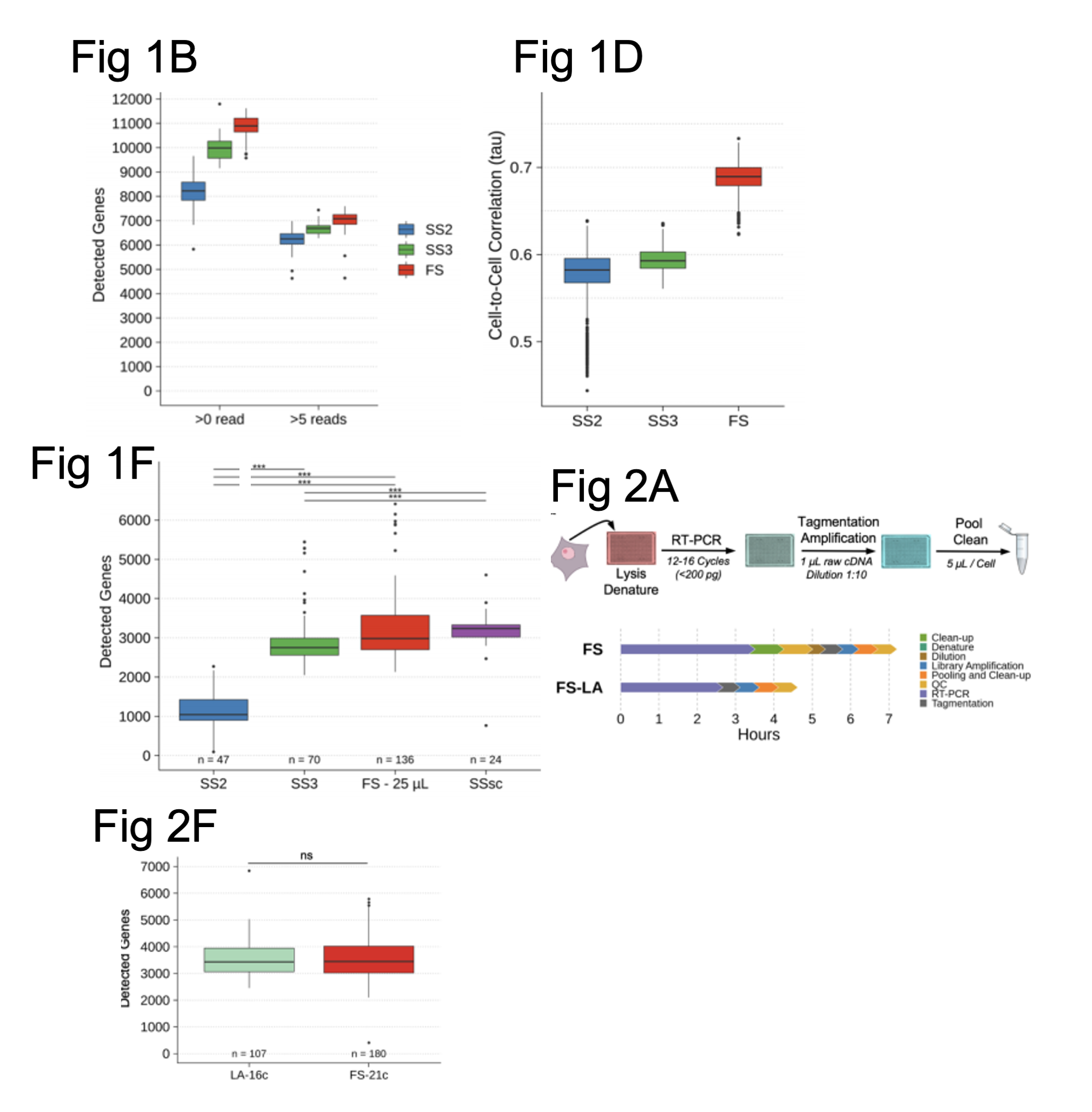
Figure shows a selection of data from Hahaut et al. Fig. 1B shows how many genes were detected using different SMART-seq protocols (SS2 and SS3) compared with FLASH-seq (FS). Fig. 1D shows the correlation between gene expression levels across SS2, SS3 and FLASH-seq protocols. Fig. 1F shows the number of genes detected in hPBMCs using SS2, SS3, a commercial single cell kit SSsc (by Takara Bio) and the FS protocol using the same volume for the final reaction. Fig. 2A. shows an illustration of the workflow for the FLASH-seq low amplification (LA) approach compared to FS. Fig. 2F compares the number of genes detected using either the FS or FS-LA approach in hPBMCs. All figures were adapted from the preprint manuscript (CC-BY-NC-ND 4.0 International license).
What I liked about this preprint:
scRNA-seq is a new and powerful tool for looking at how individual cells behave. One major barrier however is the cost in addition to the time required to prepare samples. FLASH-seq now offers an opportunity for researchers to not only prepare high quality scRNA-seq samples rapidly but it reduces the costs significantly per cell. Though such experiments are still ‘costly’, this price reduction now means scRNA-seq technology will hopefully become much more accessible to a larger community of research labs.
Questions for Authors:
Q1: What was the most challenging part of the FLASH-seq protocol to optimise?
Q2: Here you have tested FLASH-seq on human cell lines. Do you anticipate any limitations when applying this technique to other experimental organisms, for example yeasts?
Q3: Do you see any sequencing artefacts related to the increased amount of C nucleotides during the RT-PCR step?
Q4: Alternative Polyadenylation (A)A events are also important perpetrators of gene expression alterations. Can you reliably detect APA events using FLASH-seq?
Q5: Did you notice any differences in the efficacy of samples prepared using Superscript IV versus Maxima H-Reverse Transcriptase?
References:
- Zhang et al. Single-cell RNA sequencing in cancer research (2021). J. Experimental and Clinical Cancer Research
- L. Goldman et al. The impact of heterogeneity on Single-Cell sequencing. (2019). Frontiers in Genetics
- Picelli. Single-cell RNA-sequencing: The future of genome biology is now. (2017). RNA Biology
- T.P. Tang et al. Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching. (2013). Nucleic Acids Research.
doi: https://doi.org/10.1242/prelights.30332
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |