








Resolving the 3D landscape of transcription-linked mammalian chromatin folding
Posted on: 13 June 2019 , updated on: 14 June 2019
Preprint posted on 17 May 2019
Article now published in Molecular Cell at http://dx.doi.org/10.1016/j.molcel.2020.03.002
When smaller is better – Micro-C reveals transcription-linked chromatin architecture at very high resolution
Selected by Clarice HongCategories: genomics
Background
It has become well known in recent years that the 3D chromatin architecture is associated with various aspects of gene regulation. Most studies to date have focused on topologically-associated domains (TADs), which are megabase-sized regions of DNA that interact more frequently with each other than with regions outside the TAD. Due to current technical limitations, little is known about the genome architecture at smaller length scales. While TADs have been shown in some cases to influence transcriptional regulation, the relationship between the 3D genome at smaller length scales and transcription remains largely unknown. These fine scale structures include long-range enhancer-promoter interactions, which are thought to bring cis-regulatory elements such as enhancers and silencers to the gene to mediate transcription. In this preprint, the authors utilised a 3C-based technique, Micro-C (see Hsieh et al., 2015, 2016 for details), to probe chromatin organisation in mouse embryonic stem cells at much higher resolution than previous studies and understand the interplay between transcription factors/chromatin regulators and fine scale 3D genome architecture.
Key findings
After optimising the Micro-C technique for mouse embryonic stem cells, the authors first verified that the TAD organisation detected by Micro-C agrees well with Hi-C data. In another preprint that was uploaded at the same time, it was shown that Micro-C also recapitulates A-B compartments and CTCF-anchored loops with improved signal-to-noise and greater dynamic range (Krietenstein et al., 2019). In addition, Micro-C yields robust enhancer-promoter (E-P) or promoter-promoter (P-P) linkages and smaller self-interacting domains that the authors termed microTADs (Figure 1). The nucleosome-level resolution of Micro-C is also quite impressive.
Perhaps not surprisingly, many of the fine-scale structures are enriched at transcription start sites (TSSs) and intergenic regions, suggesting that they might be involved in transcriptional regulation. Boundaries of microTADs in the active compartments are enriched for CpG islands, promoters and tRNA genes and are closer to TSSs, while those in inactive compartments are enriched in repeat regions. At a finer level, boundaries are enriched for transcription factor binding sites and dynamic nucleosomes. Thus, the authors looked for factors that predict boundary strength and/or location and found a number of transcription factors, architectural proteins and histone modifications. CTCF/Cohesin are the best predictors of boundary location, while chromatin accessibility (measured by ATAC-seq) and active histone marks are the best predictors of boundary strength. Nucleosomes flanking strong boundaries also appear to be positively correlated with active histone marks and anti-correlated with repressive histone marks. Further analyses showed that microTAD boundaries can be classified into at least five non-exclusive subgroups based on biochemical/functional features – transcription-dependent (Pol II and Gro-seq), ES-cell specific enhancers (Nanog and Esrrb), constitutive enhancers (Med12 and Yy1), repressive (Ring1b and Ezh2), and insulators (CTCF and Rad21). The separation of CTCF-dependent boundaries from actively transcribing boundaries suggest that CTCF acts exclusively as a structural protein rather than a transcriptional regulator. Thus, microTAD boundaries appear to be demarcated by a combination of transcription factors and/or histone modifications and might form via various distinct mechanisms.
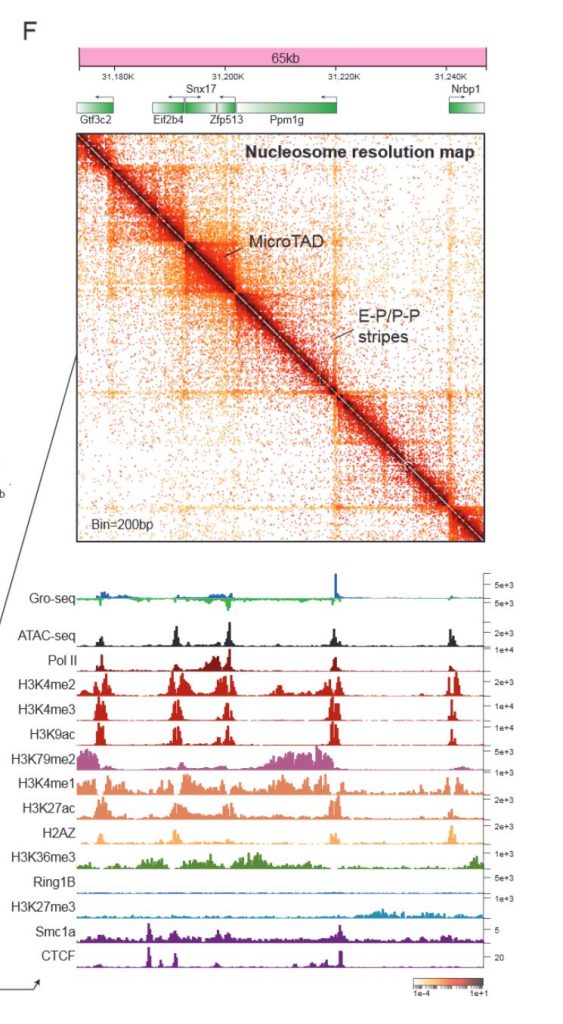
Next, the authors looked more closely at the finer-scale interactions uncovered by Micro-C. For example, P-P interactions usually extend in the same direction as Pol II elongation and are formed between co-regulated genes. E-P interactions are structurally similar to P-P interactions, and are marked by active histone modifications, Mediator, P300 and pluripotency transcription factors. The ubiquitous interactions between promoters/enhancers and other transcription factor bound loci are generally independent of CTCF/cohesin binding sites, indicating that transcription factor binding/transcription might drive the formation of such interactions. Indeed, acute inhibition of transcription did not appear to impact chromatin organisation at the scale of A/B compartments, TADs, loops or even microTAD boundaries, but reduced the strength of P-P/E-P interactions, supporting the idea that transcription drives chromatin folding at the gene level, while other mechanisms drive higher order chromatin organisation.
The nucleosome-level resolution of Micro-C also allows for the study of nucleosome packing and gene compaction. Thus, the authors looked at the relationship between transcription activity and gene compaction and found to their surprise that they are positively correlated. The authors propose two models for their finding – that Pol II might be ‘sticky’, causing actively transcribed genes to compact or that genes and their regulatory elements might be bundled in a Pol II ‘hub’, however, more experiments will need to be done to understand this somewhat counterintuitive result.
What I liked
The technique is highly impressive – it is conceptually quite a simple twist to the original C techniques but yield information at unprecedented resolution. The fact that this can now be applied to mammalian cells opens a lot of doors. There is mounting evidence that TADs and TAD boundaries often have minimal or no role in regulating transcription, and it is therefore important to look at finer-scale structures to really understand how the 3D genome might impact transcription or vice versa. Furthermore, I like the idea that there are different classes of boundaries that perhaps regulate different sets of genes, rather than the more popular idea that CTCF regulates everything from TADs to enhancer-promoter loops. Studying P-P/E-P interactions genome-wide will be so much more informative to gene regulation. The finding that transcription inhibition reduces P-P interactions and to some extent, E-P interactions suggest that the interactions are a consequence of transcription, rather than a cause. It is tempting for me to speculate that the 3D genome does not have an important role in regulating gene expression in most cases, but that it is largely a consequence of the right E-P/P-P interactions being made. Besides the actual research, I also really like that the authors have a very detailed Micro-C protocol in the supplementary materials. Sharing information is so important to pushing scientific research ahead.
Future directions and questions
It would be very interesting to test whether perturbing the predictors of microTAD boundaries has any effect of the boundaries. For example, does perturbing PolII lead specifically to loss of transcription-specific microTAD boundaries, even though it seemed to not impact boundary strength globally? Depletion of the predictors of each class would be a direct test of whether these classes are truly different from each other, and whether these are necessary for microTAD boundaries. On the other hand, it would also be interesting to see if new boundaries can be created with the insertion of TF binding sites/transcription sites to test for sufficiency.
The terminology in the preprint can also be a little confusing at times. On the one hand, at least some microTAD boundaries are enriched for promoters and are close to TSSs but are not affected by transcription inhibition. However, the E-P/P-P interactions are discussed in a separate section of the preprint and are moderately affected by transcription inhibition. Do microTAD boundaries actually overlap with these links (the model, Fig 1 above, appears to suggest that they are separate), and if so, what percentage? Are the overlaps in the first 3 microTAD boundary classes (promoters/enhancers)? Furthermore, what percentage of the boundaries cannot be explained by the five classes shown in this preprint?
References
Hsieh, T.-H.S., Weiner, A., Lajoie, B., Dekker, J., Friedman, N., and Rando, O.J. (2015). Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 162, 108–119.
Hsieh, T.-H.S., Fudenberg, G., Goloborodko, A., and Rando, O.J. (2016). Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat. Methods 13, 1009–1011.
Krietenstein, N., Abraham, S., Venev, S.V., Abdennur, N., Gibcus, J., Hsieh, T.-H.S., Parsi, K.M., Yang, L., Maehr, R., Mirny, L.A., et al. (2019). Ultrastructural details of mammalian chromosome architecture. bioRxiv 639922.
doi: https://doi.org/10.1242/prelights.11229
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Diversity and Spatial Segregation of TRP Channels in Choanoflagellates Provide Insight into the Evolutionary Origin of Animal Sensory Systems
Urvashi Goswami
BAF complexes maintain accessibility at stimulus-responsive chromatin and are required for transcriptional stimulus responses
Dina Kabbara
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |