








Selective K-ATP channel-dependent loss of pacemaking in vulnerable nigrostriatal dopamine neurons by α-synuclein aggregates
Posted on: 18 December 2019
Preprint posted on 15 November 2019
Why do alpha-synuclein aggregates affect specific neuron populations in Parkinson’s disease? a-synuclein fibrils stop pacemaking firing in PD-vulnerable midbrain dopamine neurons but not resistant neurons; K-ATP channels play a key role.
Selected by Jessica XieCategories: neuroscience
Background
One curious commonality of various neurodegenerative diseases is the issue of selective vulnerability of particular populations of neurons (Fu et al. 2018). In Parkinson’s disease (PD), there is near-total loss of dopaminergic (DA) neurons in the lateral substantia nigra (SN) of the midbrain, while DA neurons in the medial SN and ventral tegmental area (VTA) are significantly less affected (Surmeier et al. 2017; Kordower et al. 2013). Huntington’s disease initially affects striatal medium spiny neurons projecting to the external globus pallidus, while sparing those projecting to the internal globus pallidus. Alzheimer’s disease notably impacts the hippocampus and entorhinal cortex but not motor and sensory cortex; ALS too differentially affects various motor neuron populations.
This feature of selective neuronal vulnerability is an intriguing one, not just in terms of biology (what differentiates subtypes of neurons?), but also for its potential therapeutic significance: discovering what underlies the vulnerability—or conversely, resistance—of certain neuronal subtypes to the disease-causing process would be a major step towards understanding the disease, and consequently developing strategies for its treatment or prevention.
How then to identify the key factors distinguishing neuronal subtypes? Several previous studies in the field of PD have compared expression profiles of SN and VTA and proposed potential area-specific markers (notable mentions include GIRK2 and CALB1), but to date there are no definitive molecular markers of SN or VTA regional identity (Anderegg et al. 2015; Brichta et al. 2014). Some electrophysiological differences are better established—for example, pacemaking in SN DA neurons is understood to be driven by voltage-gated calcium channels, and pacemaking in VTA DA neurons by voltage-gated sodium channels (Chan et al. 2007). Additionally, the Roeper lab has previously identified activation of K-ATP channels in SN but not VTA, and suggested that this could be responsible for selective SN DA neuronal death (Liss et al. 2005).
In addition to selective neuronal death, many neurodegenerative diseases are also associated with aberrant protein aggregation in affected regions—PD, for example, is characterized by Lewy body aggregates composed primarily of the a-synuclein protein. While it is widely accepted that a-synuclein is critically involved in PD pathogenesis, how exactly is unclear. It has been suggested that the process of a-synuclein aggregation and/or Lewy body formation may be stressful and consequently toxic, but could fibrils themselves be acutely toxic? Are pathogenic fibrillar forms of a-synuclein preferentially produced in the vulnerable region of the midbrain, or are these a-synuclein fibrils only toxic to certain midbrain neurons, but not others—and if so, what molecular differences underlie this disparity?
This study by Thakur et al. provides the tempting beginnings of a potential resolution to these important outstanding issues in the field of PD, with the exciting finding that a-synuclein fibrils cause dramatic loss of pacemaking firing in regions vulnerable in PD, while having a much smaller effect in other neighboring, resistant regions—and that K-ATP channels could potentially be the reason why.
Key findings
The authors began by performing retrograde tracing in order to label subpopulations of neurons within the mouse midbrain, taking advantage of the fact that selectively vulnerable and resistant midbrain neuron populations can be anatomically identified. Specifically, they injected fluorescent beads into the dorsolateral striatum, innervated by lateral SN DA neuron populations most vulnerable in PD. They also made injections into the dorsomedial striatum and ventral striatum (nucleus accumbens), labeling less affected midbrain DA neurons in the medial SN and VTA.
Next, the authors applied a-synuclein fibrils intracellularly to these labeled neurons, by performing whole-cell patch clamp electrophysiology with a-synuclein fibrils added to the internal recording solution (which theoretically equilibrates with cellular contents). While a-synuclein fibrils are generally applied extracellularly (in cell culture media or through in vivo injections) and as a chronic treatment lasting days to months (Luk et al. 2012; Volpicelli-Daley et al. 2011), this relatively novel experimental paradigm—intracellular application of a-synuclein fibrils—resulted in a remarkable finding: a-synuclein fibrils markedly and rapidly disrupted the endogenous pacemaking activity of midbrain DA neurons, causing a complete loss of pacemaking firing activity in a majority (65%) of lateral SN neurons within 5 minutes. Moreover, this deficit was specific to the PD-vulnerable midbrain DA neuron population, namely lateral SN neurons projecting to dorsolateral striatum. In contrast, medial SN and VTA neurons innervating dorsomedial and ventral striatum were significantly less affected.
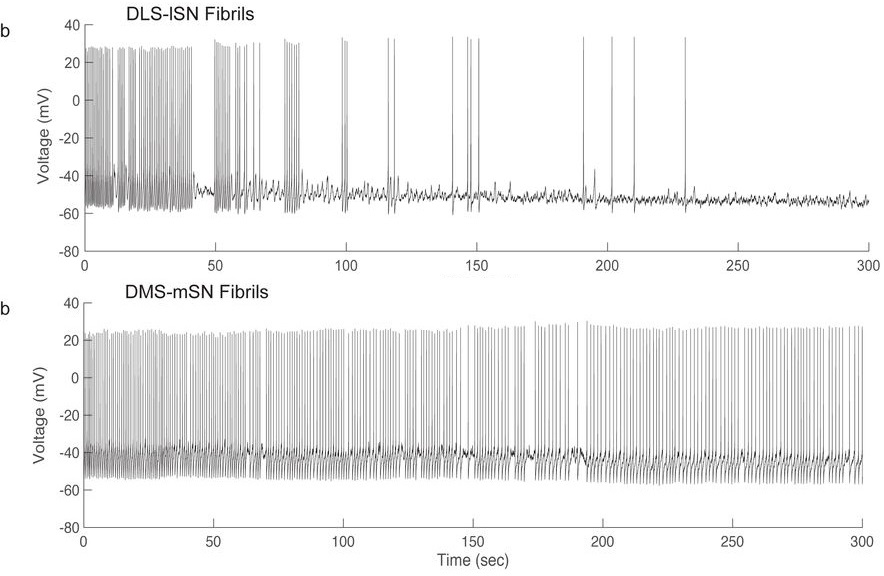
The authors additionally showed that monomeric forms of a-synuclein had a reduced effect on the pacemaking activity of lateral SN DA neurons, consistent with many previous studies showing greater pathological effects of fibrillar forms of a-synuclein relative to monomers. Furthermore, extracellular application of a-synuclein fibrils also acutely interfered with pacemaking activity, confirming that this was not an experimental artifact of intracellular a-synuclein fibril application.
But what mechanisms might underlie this regional vulnerability? The authors show that bath application of a K-ATP channel inhibitor, glibenclamide, partially protected against the detrimental effect of a-synuclein fibrils, suggesting a possible mechanism that also nicely corroborates their previous work on SN K-ATP channels (Liss et al. 2005).
Questions for the authors:
- I was intrigued by your experimental choice to look at the acute effect of a-syn fibril application, as most studies seem to go chronic/long-term. Any additional backstory to share behind your decision to investigate the acute effect on an ephys feature?
- I’m also very impressed by how fast and dramatic the reported finding was, especially given that most PD/a-syn fibril phenotypes have been quite slow/subtle (e.g. takes weeks for some cell death). Upon first read I’d initially assumed it must be because of intracellular application, then realized that extracellular fibrils also had a rapid effect on timescale of seconds. I’m wondering if this could be a potential clue as to mechanism: might you speculate if it could be a-syn fibril binding to extracellular region of protein (K-ATP channel maybe?), some rapid protein uptake/import, or something else?
- Finally, could you speculate how loss of pacemaking activity might lead to neuronal death/neurodegeneration?
References
- Anderegg, A., Poulin, J.F., & Awatramani, R. (2015). Molecular heterogeneity of midbrain dopaminergic neurons—Moving toward single cell resolution. FEBS letters, 589(24 Pt A), 3714-26. https://doi.org/10.1016/j.febslet.2015.10.022
- Brichta, L., & Greengard, P. (2014). Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: an update. Frontiers in neuroanatomy, 8, 152. https://doi.org/10.3389/fnana.2014.00152
- Chan, C.S., Guzman, J.N., Ilijic, E., Mercer, J.N., Rick, C., Tkatch, T., … & Surmeier, D.J. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature, 447(7148), 1081-6. https://doi.org/10.1038/nature05865
- Fu, H., Hardy, J., & Duff, K.E. (2018). Selective vulnerability in neurodegenerative diseases. Nature neuroscience, 21(10), 1350-8. https://doi.org/10.1038/s41593-018-0221-2
- Kordower, J.H., Olanow, C.W., Dodiya, H.B., Chu, Y., Beach, T.G., Adler, C.H., … & Bartus, R.T. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain : a journal of neurology, 136(Pt 8), 2419-31. https://doi.org/10.1093/brain/awt192
- Liss, B., Haeckel, O., Wildmann, J., Miki, T., Seino, S., & Roeper, J. (2005). K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nature neuroscience, 8(12), 1742-51. https://doi.org/10.1038/nn1570
- Luk, K.C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J.Q., & Lee, V.M. (2012). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science (New York, N.Y.), 338(6109), 949-53. https://doi.org/10.1126/science.1227157
- Surmeier, D.J., Obeso, J.A., & Halliday, G.M. (2017). Selective neuronal vulnerability in Parkinson disease. Nature reviews. Neuroscience, 18(2), 101-13. https://doi.org/10.1038/nrn.2016.178
- Volpicelli-Daley, L.A., Luk, K.C., Patel, T.P., Tanik, S.A., Riddle, D.M., Stieber, A., … & Lee, V.M. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 72(1), 57-71. https://doi.org/10.1016/j.neuron.2011.08.033
doi: https://doi.org/10.1242/prelights.15883
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the neuroscience category:
Behavioral characteristics of an extremely old rhesus macaque in a zoo: Dementia-like symptoms and implications for quality of life of geriatric animals
Stefan Friedrich Wirth
EBV reprograms autoreactive anti-CNS B cells as antigen presenting cells in multiple sclerosis
Léa Bastien et al.
The Endocannabinoid System’s Contribution to Placebo Analgesia
Thomas Nicodemo Arrieta et al.
preLists in the neuroscience category:
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
SDB 78th Annual Meeting 2019
A curation of the preprints presented at the SDB meeting in Boston, July 26-30 2019. The preList will be updated throughout the duration of the meeting.
| List by | Alex Eve |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Young Embryologist Network Conference 2019
Preprints presented at the Young Embryologist Network 2019 conference, 13 May, The Francis Crick Institute, London
| List by | Alex Eve |