








Simultaneous multiplexed amplicon sequencing and transcriptome profiling in single cells
Posted on: 20 November 2018
Preprint posted on 13 November 2018
Article now published in Nature Methods at http://dx.doi.org/10.1038/s41592-018-0259-9
High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes
Posted on:
Preprint posted on 24 September 2018
Article now published in Nature Communications at http://dx.doi.org/10.1038/s41467-019-11049-4
What’s all the RAGE: DARTing into uncharted droplet-based scRNA-seq territory
Selected by Samantha SeahCategories: genomics
Background
Droplet-based single-cell RNA sequencing (scRNA-seq) is, and continues to be an important tool for estimating changes in gene expression at a single-cell level in a high-throughput manner. However, current techniques are largely limited to studying the 3’ ends of A-tailed mRNA transcripts at a transcriptome-wide level.
These two preprints by Saikia and Burnham et al. (on DART-seq) and Singh and Al-Eryani et al. (on RAGE-seq) tackle various limitations of current droplet-based scRNA-seq. For example, the diversity of B- and T- cell receptors (BCRs and TCRs respectively) generated by V(D)J recombination, and the further random addition or removal of nucleotides cannot be elucidated by traditional droplet-based scRNA-seq. The V(D)J regions of the B- and T-cell receptors are located at the 5’ end of their respective transcripts, while the combination of 3’ end capture, fragmentation and short-read Illumina sequencing in droplet-based scRNA-seq results in one getting sequences largely restricted to the 3’ end. The two preprints both tackle this problem, albeit slightly differently – DART-seq uses specific primers and modified Drop-seq beads to capture the heavy and light chain transcripts just downstream of the variable region, while RAGE-seq uses Oxford Nanopore technology to sequence the captured full-length BCR and TCR transcripts.
Additionally, Saikia and and Burnham et al. used DART-seq to characterise non-A-tailed transcripts of dsRNA viruses, while Singh and Al-Eryani et al. used RAGE-seq to characterise alternative splicing and gene rearrangements in BCR transcripts, both of which would be extremely difficult with traditional droplet-based scRNA-seq.
DART-seq (Saikia and Burnham et al.)
In DART-seq, customised primers are ligated to a fraction of poly-dTs on Drop-seq beads, with an efficiency of 25-40%, which allows the specific capture and amplification of transcripts of interest. The ligation reaction can be titrated to leave many poly-dTs available, enabling the simultaneous capture of transcripts-of-interest and A-tailed transcripts.
The authors use their technology to specifically capture and sequence heavy and light chain antibody variable regions, while simultaneously obtaining whole-transcriptomic data from those same cells. Antibody variable regions are located at the 5’ end of their transcripts and traditionally cannot be sequenced with droplet-based scRNA-seq, which focuses on the 3’ end of transcripts. By designing primers just downstream of the V(D)J segments, the Ig recovery rate could be increased significantly compared to Drop-seq.
DART-seq was then used to study the B-cell antibody repertoire within human peripheral blood mononuclear cells (PBMCs). Of the 818 B-cells identified, 564 (67%) had immunoglobulin transcripts and the complete heavy and light chain CDR3 regions were obtained for 120 cells (15%). Clone-specific pairing was measured, and the highest frequency was observed between the most highly expressed heavy and light chain transcripts, which supports previous data.
The authors also applied their technology to study the infection of cells with T3D reovirus, an RNA virus with non-A-tailed transcripts. Infected cells were subjected to DART-seq, with beads modified to capture segments of the viral genome. By using primers designed to sequence the entire S2 transcript, the authors were able to analyse point mutations in the viral transcripts. Upon study of the cell transcriptomes, they found four distinct cell subpopulations, one of which was not found in the non-infected control.
RAGE-seq (Singh and Al-Eryani et al.)
In contrast to DART-seq, RAGE-seq uses targeted capture coupled with long-read sequencing with Oxford Nanopore. To obtain full-length BCR and TCR sequences, a capture bait library targeting all V, J and constant (C) region exons was used. The cDNA library is simultaneously sequenced via traditional short-read sequencing (via 10X Genomics), enabling 3’ expression profiling.
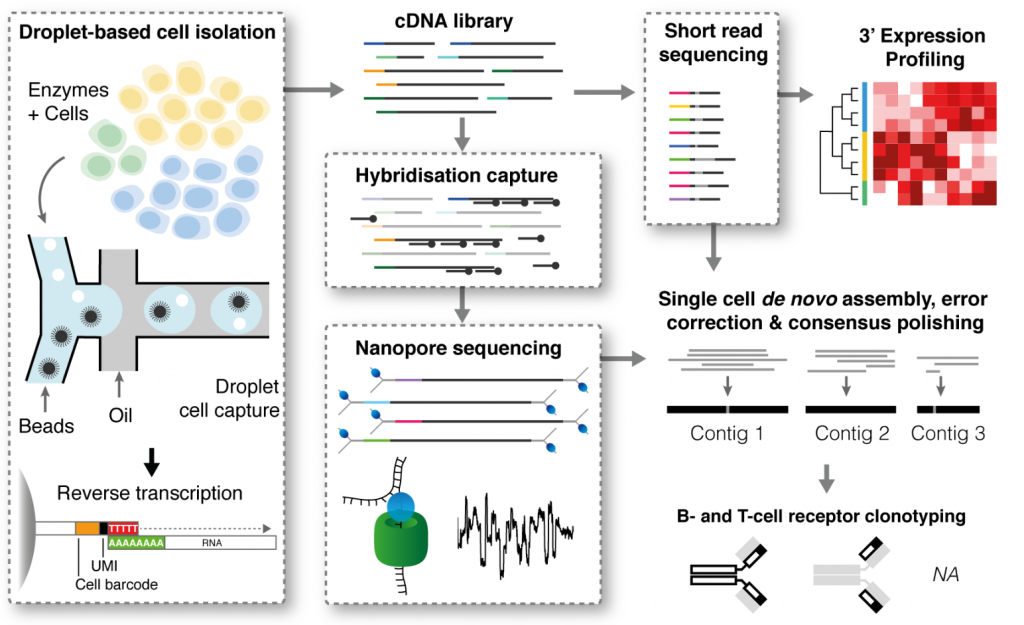
Figure 1 of Singh and Al-Eryani et al., made available under a CC-BY-NC-ND 4.0 International License
The authors validated the technology with a mix of Jurkat (T cells), Ramos (B cells) and monocytes (as a negative control). They noted a 13-fold enrichment of nanopore reads aligned to the BCR and TCR, when compared to non-targeted capture, and were able to recover both full length transcripts from a subset of cells – TCRα and β from 18.9% of Jurkat cells and the Ig heavy and light chains from 31% of Ramos cells. By tracking the amino acid mutations present, the authors could plot and follow the evolution of Ramos cells undergoing somatic hypermutation.
The authors then analysed lymphocytes from a human lymph node, where they similarly identified T cells and B cells, and sequenced their respective TCR and immunoglobulin chains. They quantified differences in Ig heavy chain transcripts in different cell types, noting that naive and memory B cells have both membrane and secretory IGH isoforms, while plasmablasts and plasma cells only have the secretory form. In addition, many plasmablasts and plasma cells were assigned to IGHA1, which is consistent with differentiation to high-rate antibody secreting cells.
The lymph node analysis was combined with analysis of lymphocytes from the primary breast tumour of the same patient, to enable the tracking of clonally-related T and B cells across different tissues. 7 clones were found to be shared between the tumour and lymph node, of which 6 were found within the CD8 T cell cluster. These clonal expanded cells have a discrete gene signature associated to active tissue resident cytotoxic lymphocytes, but each clone also seemed to express unique sets of genes.
What I like about these works
High throughput droplet-based transcriptomic sequencing is an extremely powerful technique, but currently suffers from certain limitations, such as the inability to sequence nucleotides located at the 5’ end of transcripts and the lack of sequencing strategies to pick up non-A-tailed transcripts.
The developments proposed by Saikia and Burnham et al. and Singh and Al-Eryani et al. go a long way to solve these problems. The two groups of authors have come up with innovative and contrasting technologies that expand the single-cell transcriptomic toolkit. In particular, both preprints have demonstrated an ability to recover natively-paired heavy and light chains of antibody variable regions, which is difficult to obtain with traditional droplet-based single-cell transcriptomic sequencing.
I’m also enthralled by the recent increase in utilisation of Oxford Nanopore sequencing, and I find the combination of Oxford Nanopore and Illumina sequencing in RAGE-seq (Singh and Al-Eryani et al.) exciting and potentially useful for many applications.
doi: https://doi.org/10.1242/prelights.5740
 (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |