








Expressive modeling and fast simulation for dynamic compartments
Posted on: 18 April 2024 , updated on: 31 March 2025
Preprint posted on 3 April 2024
Article now published in PLOS One at https://doi.org/10.1371/journal.pone.0312813
Running dynamic compartment mathematical models faster and directly in your browser with the third generation of the multi-level modelling and simulation framework (ML-Rules).
Selected by Benjamin Dominik MaierCategories: bioinformatics, systems biology
Updated 31 March 2025 with a postLight by Benjamin Maier
Congratulations to Till Köster, Philipp Henning, Tom Warnke, and Adelinde Uhrmacher on the publication of their manuscript in PLOS One! In the journal version, the key contributions are more explicitly outlined, and a couple of sentences/short paragrpahs have been added throughout the manuscript to more clearly reason their approach and/or add additional context.
Background: Rule-based Modelling
Biomolecules like proteins typically have multiple post-translational modification sites and binding partners. Traditional modelling approaches individually define reactions for each possible interaction, which quickly becomes unfeasible considering their combinatorial complexity (Chylek et al., 2015). When looking at a protein with just three phosphorylation sites (○) which can be phosphorylated (●), 12 reactions are needed to describe all possible states explicitly (Fig. 1, left). However, employing implicit reaction templates in rule-based modelling reduces this to just 3 patterns (Fig. 1, right). For instance, the pink rule states that if the left phosphorylation site is unphosphorylated, it can be phosphorylated. This pattern can then be applied to input states fulfilling these defined contextual prerequisites to update their state whenever they are selected. For proteins with more sites and binding partners like the epidermal growth factor receptor (EGFR) complex the difference becomes even more striking, as 1.9×108 states can be described implicitly with just 20 reactions (Blinov et al., 2006). More information about rule-based modelling can be found in a review paper from Chylek et al. (2013).
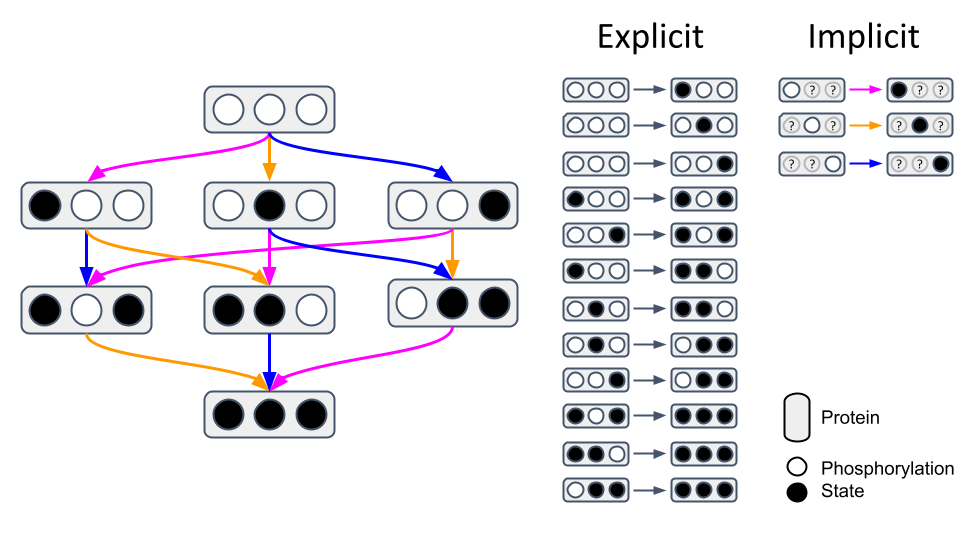
Fig. 1: Explicit vs. implicit network representation. Figure created with PowerPoint.
Key Findings
In this preprint, Köster and colleagues built on previous work and have improved their multi-level modelling and simulation framework of cellular systems (ML-Rules) (Maus et al. 2011; Warnke et al., 2015). Their study aimed to 1) account for flexible modelling of compartmental dynamics, 2) optimise the performance of the simulation routine with a new simulation engine and 3) enable web browser-based simulations.
Dynamic Compartments
Most modelling frameworks consider reactions confined to specific compartments, with their reaction dynamics influenced by factors like compartmental volume, temperature, pH, and reactant density. However, they typically treat compartments as static entities, simplifying simulations but preventing modellers to realistically represent structural compartment changes like fission/fusion, formation/disintegration and translocation processes (Fig. 2). In 2011, John et al. proposed React(C) to express dynamic compartments through hyperedges (concept outlined in Suppl. Fig. 1). Yet no hypergraph-rewriting-based simulator exists due to computational complexity and model specification challenges.
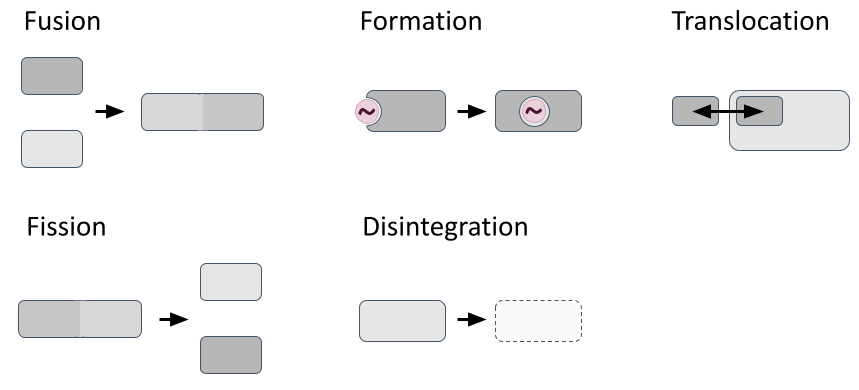
Fig. 2: Dynamic structural compartment changes. Figure created with PowerPoint.
ML-Rules version 3 used in this study is based on an alternative approach called multiset term rewriting, where the model is represented by n-ary trees (see GeeksForGeeks Tutorial and slides by Paolo Milazzo). Let us take a three species system S={A, B, C } with multiset rewriting rules {AB → C , C → AB}, we can compute its traces in advance; e.g. A5B3 → A4B2C → A3BC2 → A2C3 → A3BC2 → A4B2C → A5B3 and then just apply these operations on those multiset of entities involved whenever needed.
Performance Updates
As part of this study, ML-Rules has been re-implemented in the Rust programming language to improve its performance. Additionally, a new hybrid rule-based simulation method has been developed, allowing individual attributes to be simulated in a network-free manner: Following consistency checks, all possible species and reactions are enlisted in a flat representation to speed up the actual simulation (in-advance enumeration). Attributes that cannot be easily represented in a network-based manner are not explicitly enumerated before simulation execution but instead instantiated with appropriate values only once the actual rule is selected and applied. After some optimization steps, the model is simulated using the stochastic simulation algorithm (SSA) (outlined in my previous preLights post). Whenever structural compartment changes occur (see Fig. 2), the model is transformed back into a tree-like hierarchical representation to apply them before reinitiating the routine.
Web Server-based Simulations
ML-Rules 3 comes with a web server for running model simulations directly in the browser, eliminating the need for software installation or browser extensions. Executable models can be shared and executed using custom URLs, which enhances accessibility and reproducibility. Unlike other web server-based simulation tools, ML-Rules 3 operates directly on the user’s machine, cutting costs associated with server or cloud systems.
Case Study I: Fission Yeast Model
In order to show the improved performance of ML-Rules 3, Köster and colleagues turned to their multi-cellular model of fission yeast from Maus et al. 2011. This model describes the yeast cell cycle, mating type switching, cell cycle inhibition mechanisms between cells of opposite mating type as well as cell division (Fig. 3) and consists of 15 rules. Simulating the model with ML-Rules 3 was shown to be 100x faster than the old Java-based implementation of ML-Rules 2. While running the simulation in the web browser (link to run it yourself) slows down the simulation slightly, it remains more than 50x faster.

Fig. 3 Fission Yeast Model from Maus et. al (2011). Figure taken from Köster et al. (2024), BioRxiv published under the CC-BY 4.0 International licence.
Case Study II: mRNA Delivery Model
Next, Köster and colleagues revised a previously published multi-level kinetic model of mRNA delivery via the transfection of small lipid spheres containing mRNA (so called lipoplexes) (Ligon et al., 2014) to demonstrate the improved versatility of ML-Rules 3 regarding dynamic compartments. In the model, extracellular lipoplexes attach to the cell surface to enter the cell via endocytosis (Fig. 4). This leads to the formation of endosomes inside the cell which can either lyse or degrade. If they lyse, the internal lipoplex can release their mRNA content into the cell. Subsequently, the mRNA can undergo translation within the cell, leading to protein synthesis. Meanwhile, endosomes, lipoplexes, mRNA, immature as well as mature proteins are constantly degraded.
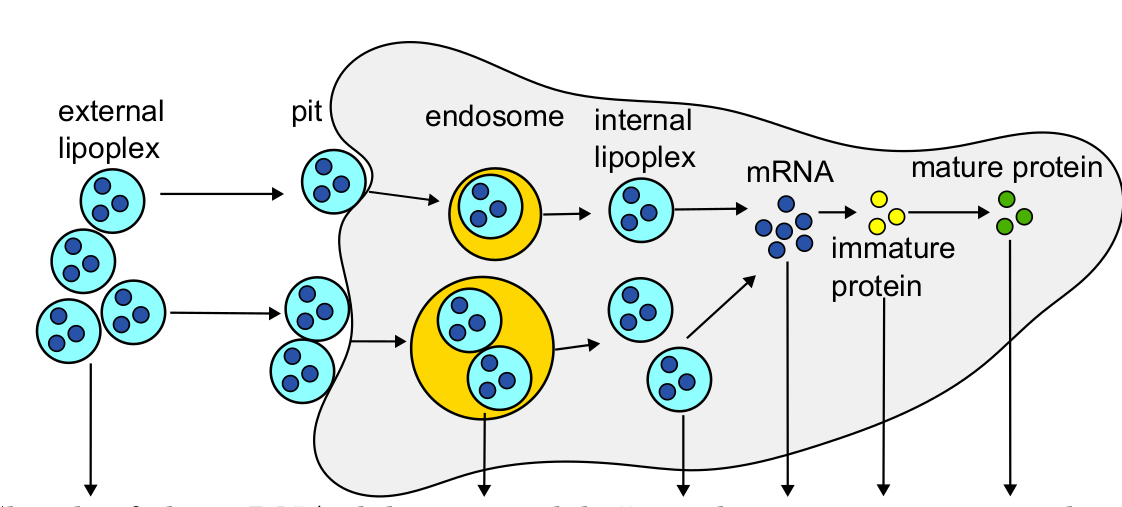
Fig. 4 Lipoplex Transfection Model from Ligon et al. (2014). Figure taken from Köster et al. (2024), BioRxiv published under the CC-BY 4.0 International licence.
Ligon and colleagues had to make multiple simplifications to make their COPASI model implementation and simulation feasible. These include setting a fixed number of mRNA molecules per lipoplex despite experimental data showing large variations and not accounting for lipoplexes unpacking their mRNA directly into endosomes where it is degraded. In the reimplemented model presented in this preprint, the lipoplexes can be treated as individual dynamic compartments allowing for varying mRNA amounts as well as the unpacking of mRNA into the endosome for degradation. This thereby overcomes the two key limitations of the earlier model. When re-running the model, the authors found that it behaved more realistically and in better accordance with the experimental wet-lab data. Moreover, the simulation time was shown to be 6x faster in the ML-Rules 3 implementation compared to the previous COPASI implementation.
References
Blinov ML, Faeder JR, Goldstein B, Hlavacek WS. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. Biosystems. 2006;83(2):136-151. https://doi.org/10.1016/j.biosystems.2005.06.014
Chylek, L. A., Harris, L. A., Tung, C. S., Faeder, J. R., Lopez, C. F., & Hlavacek, W. S. (2014). Rule-based modeling: a computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley interdisciplinary reviews. Systems biology and medicine, 6(1), 13–36. https://doi.org/10.1002/wsbm.1245
Chylek, L. A., Harris, L. A., Faeder, J. R., & Hlavacek, W. S. (2015). Modeling for (physical) biologists: an introduction to the rule-based approach. Physical Biology, 12(4), 045007. https://doi.org/10.1088/1478-3975/12/4/045007
John, M., Lhoussaine, C., Niehren, J., & Versari, C. (2011). Biochemical Reaction Rules with Constraints. Programming Languages and Systems, 338–357. https://doi.org/10.1007/978-3-642-19718-5_18
Ligon, T. S., Leonhardt, C., & Rädler, J. O. (2014). Multi-Level Kinetic Model of mRNA Delivery via Transfection of Lipoplexes. PLOS ONE, 9(9), e107148. https://doi.org/10.1371/journal.pone.0107148
Maus, C., Rybacki, S., & Uhrmacher, A. M. (2011). Rule-based multi-level modeling of cell biological systems. BMC Systems Biology, 5(1), 166. https://doi.org/10.1186/1752-0509-5-166
Warnke, T., Helms, T., & Uhrmacher, A. M. (2015). Syntax and Semantics of a Multi-Level Modeling Language. Proceedings of the 3rd ACM SIGSIM Conference on Principles of Advanced Discrete Simulation, 133–144. Presented at the ACM SIGSIM Conference London, United Kingdom. https://doi.org/10.1145/2769458.2769467
Supplementary Materials
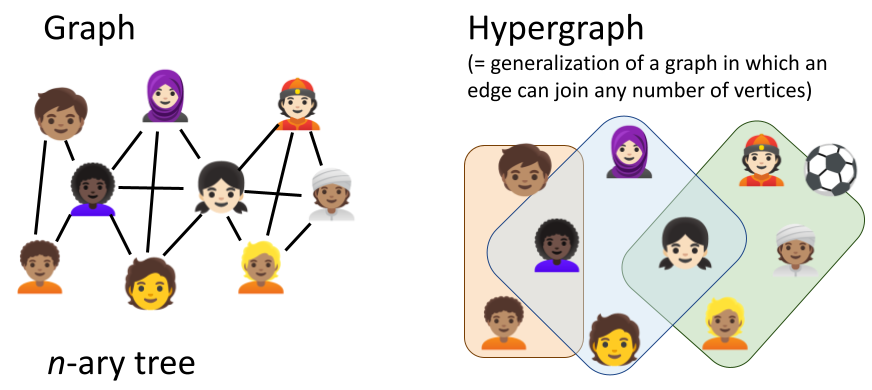
Suppl. Fig. 1 Difference between graphs and hypergraphs in expressiveness. Unlike traditional binary graphs (either two nodes/vertices are connected by an edge or not), hypergraphs allow multi-way connections between any number of vertices. Figure created with PowerPoint.
doi: https://doi.org/10.1242/prelights.37158
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
The lipidomic architecture of the mouse brain
CRM UoE Journal Club et al.
Kosmos: An AI Scientist for Autonomous Discovery
Roberto Amadio et al.
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Also in the systems biology category:
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
Single-Cell Network Analysis Identifies CLEC4E as a Key Mediator of Proinflammatory mDC Responses in Influenza Infection
Charis Qi
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |