








Digitize your Biology! Modeling multicellular systems through interpretable cell behavior
Posted on: 8 November 2023
Preprint posted on 24 September 2023
Article now published in Cell at http://dx.doi.org/10.1016/j.cell.2025.06.048
From Lab to Model: Lowering the entry barriers into mathematical modelling with an intuitive human-readable modelling language
Selected by Benjamin Dominik Maier, Lucy Bowden, Jennifer Ann Black, Saanjbati AdhikariIntroduction
When starting a new project in the lab, the first step is usually to study the existing biological knowledge which leads to the formulation of a hypothesis that can be empirically tested. Next, pilot experiments are designed and performed to find the best conditions and experimental setup to answer the chosen research question. The experiments are then conducted and the resulting data is analysed and interpreted to gain new insights and answer the research question.
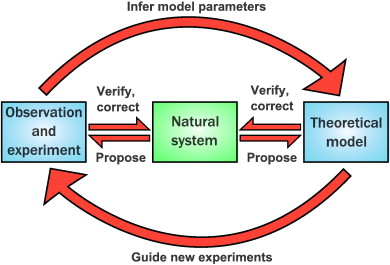
Figure 1: Interaction between experimental and computational biology experiments. Figure taken from Székely, T., Jr, & Burrage, K. (2014), Computational and structural biotechnology journal, published under the CC BY 3.0 Deed licence.
While most of these steps could benefit from complementary in silico analysis, this is rarely done, often due to a lack of expertise and the high entry barrier to bioinformatics and systems biology approaches (see section below). However, the use of mathematical modelling can help researchers make predictions towards which parameters are most important to measure. It thereby can help to identify the optimal set of conditions that are likely to yield meaningful results. As such, the number of experimental trials needed to test a given hypothesis can be reduced saving both time and resources. Besides this, mathematical models can be constructed based on the experimental measurements to simulate a large number of different conditions. This can help researchers pinpoint knowledge gaps, gain new insights and generate new hypotheses to experimentally explore and validate.
The preprint discussed here proposes a new conceptual framing (a “grammar”) for specifying cell behaviour hypotheses – one that can be used and interpreted not just by computational biologists, but by the scientific community at large. The authors demonstrate the simpleness and versatility of their human-readable conceptual framing with five examples of varying complexity and show how model parameters can be directly inferred from spatial transcriptomics data and translated into agent-based models.
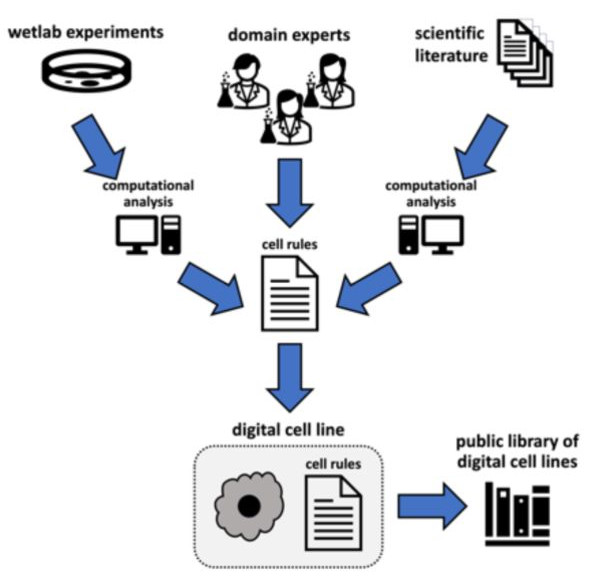
Figure 2: Agent-based Modelling Workflow. Figure taken from Johnson, Stein-O’Brien et al. (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Identifying Mathematical Modelling Entry Barriers
Experimentalists are facing numerous entry barriers when it comes to modelling – the biggest one probably being how and where to start? Moreover,
- Implementing mathematical models often requires knowledge of advanced mathematical/statistical concepts and proficiency in programming languages. Learning and applying these methods is time-consuming, particularly for people who are not already familiar with the field.
- Mathematical models often use technical language and formats that demand a level of proficiency in understanding and interpreting the definitions and notations within the model.
- There is a lack of interdisciplinary communication and collaboration with systems biologists and mathematicians.
- Making an informed decision about the most suitable modelling framework (boolean, ODE, agent-based, rule-based) requires a lot of modelling expertise
- Developing mathematical models that capture the relevant biological processes while remaining mathematically tractable can be difficult.
- Running simulations and analyses on complex models may require access to high-performance computing resources, which may not be readily available.
Who we are and how mathematical modelling/the presented modelling framework might aid our projects?
Lucy Bowden: I am a microbiologist focusing on bacterial attachment and biofilm growth. I think that a modelling framework such as this one would be very interesting in my work if it could help me model bacterial community growth on varied substrates given different mechanical parameters, as well as in different environmental conditions.
Jennifer Black: I am a parasitologist working on DNA repair and genome plasticity in the parasite Leishmania. A modelling system like this could be valuable in my area if it could be used to understand how parasites may respond when faced with stressful conditions.
Saanjbati Adhikari: I am a biochemist and cell biologist, focussing on chromosome segregation mechanisms during mitosis in mammalian cells. I believe that a modelling approach like the one presented here can be valuable in predicting the dynamics of key players in cell division, including spindle microtubules and kinetochores, under both normal and diseased/ stressful conditions.
Benjamin Maier: I am a systems biologist working on the development of context-specific modelling workflows to study cell signalling and signal rewiring. This preprint, along with the discussions among us preLighters, inspired me to think about how to make my own modelling frameworks more accessible to people outside the community and provided me with some new ideas and concepts. Moreover, it encouraged me to reflect about how we can foster greater exchange and interdisciplinarity between experimental and computational biologists.
Perspective on the Manuscript
What we liked:
- When we read this preprint, we were struck by the ease with which we could understand the described input commands (“grammar”). For those of us who are not computational biologists, this significantly lowers the barriers to our participation in and understanding of mathematical modelling.
- We were also intrigued by the way this project is set up: the method presented is meant to be community-driven, with the ideal that future developments will be driven by actual need.
- We were impressed by the multiple examples included in this preprint, which naturally progressed from a more simple to a rather complex model. We can see how the presented modelling approach can have a wide variety of applications in different subdisciplines of biology.
- We appreciated how the authors managed to integrate experimental data directly into their models (here: bulk and single-cell RNA-seq and spatial transcriptomics). This makes it even more clear to experimental biologists how using the computational method presented here can benefit their work directly by allowing for personalised predictions.
What we were wondering:
- We asked ourselves whether the presented model is so simplified that it fails to maintain relevance (especially from the perspective of those already involved in the modelling community).
- In order to showcase the versatility of the modelling approach, we believe that the paper would be strengthened if the examples came from several biological fields rather than focusing only on cancer immunology.
- Although it is clear that the presented modelling approach makes important steps to increase accessibility and the adoption by the larger community, how can one ensure that users are properly trained/guided to use this resource responsibly. Also, we were wondering how much initial modelling experience one needs to use this resource.
Our wishlist
- We hope that proactive efforts will be made to get people both within and outside (!) of the computational biology community to use this resource.
- We would like to see an example where this modelling approach is used in the context of research on prokaryotes (though ideally for more complex organisms and systems such as biofilms of non-model organisms)
- To show the wide range of applications for this computational tool, perhaps a (community-driven) repository and vignette with examples can be created. This could help modelling novices to start their first project and get inspirations from existing models.
- We like the idea of adding AND and REQUIRES operators to describe relationships as well as having a negation symbol. Nevertheless, we recognize that with an expanding toolbox, the language might not be as accessible and intuitive to new users.
Questions to the authors
Q1: How can you make sure that all relevant parameters are included when trying to model a complex biological system? And expanding on that, how can you make sure that the parameters you model are indeed relevant?
Q2: Does the fact that the computational model aligns with experimental results necessarily prove the model’s validity? What are the criteria for this?
Q3: In example 4, you state that “these results show a highly successful anti-tumor response in terms of T cell activation and expansion, but complete clearance of the tumour core is not achieved by the simulation endpoint.” Have you run the model with more iterations and if so, do you observe a complete clearance at a later time point?
Q4: The discussion describes plans to consult with the community – how will you expand to people outside the computational biology community? How can you involve as many people as possible in this discussion?
Q5: Even though it is possible to express the statements “low oxygen increases necrosis” and “oxygen decreases necrosis” with a shared mathematical expression, they convey
different meanings and potentially address different research hypotheses. In general, how can we ensure that the model rules are reflective of the biological system and rule out that subtle differences in phrasing significantly affect the results of a model?
doi: https://doi.org/10.1242/prelights.35819
Read preprint (1 votes)
(1 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the scientific communication and education category:
Science should be machine-readable
Theodora Stougiannou
A thirty-year trend of increasing clinical orientation at the National Institutes of Health
AND
Prediction of transformative breakthroughs in biomedical research
Jonathan Townson
DNA Specimen Preservation using DESS and DNA Extraction in Museum Collections: A Case Study Report
Daniel Fernando Reyes Enríquez, Marcus Oliveira
Also in the systems biology category:
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the scientific communication and education category:
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |