








Inhibitory regulation of calcium transients in prefrontal dendritic spines is compromised by a nonsense Shank3 mutation
Posted on: 27 February 2020
Preprint posted on 8 January 2020
Article now published in Molecular Psychiatry at http://dx.doi.org/10.1038/s41380-020-0708-6
Don’t forget the interneurons! Ali et al. find reduced dendritic inhibition by Shank3-mutant interneurons in the prefrontal cortex is to blame for dendritic calcium dysregulation.
Selected by Osvaldo A. MirandaCategories: neuroscience
Introduction:
The basic computational units of the brain are glutamatergic and GABAergic neurons, which work together to process and relay data in a coherent manner. This cooperation relies on striking a fine balance between excitation and inhibition, too much of one or the other is thought to lead to neurological disorders such as autism spectrum disorder (ASD), epilepsy, and schizophrenia1,2,3. Many studies on ASD focus heavily on the effects of mutation on pyramidal neurons (PNs), while a smaller fraction place their focus on interneurons (INs). One high-profile ASD candidate gene is Shank3, a gene encoding a post-synaptic scaffold protein, which when mutated disrupts synapse formation and maturation in PNs and may lead to ASD, schizophrenia or Phelan-McDermid syndrome. Numerous studies have outlined though its effects on INs remain poorly understood. In this preprint, Ali et al. give some attention to the minority cell types of the cortex, the good ole IN. Specifically, they highlight the effects of Shank3 mutations on IN function and how this in turn results in behavioral phenotypes analogous to symptoms of ASDs.
Key findings:
Elevated synaptic calcium transients in prefrontal cortex of R1117X mice
To study the effects of Shank3 mutations on cortical circuitry the authors use R1117X knock-in mice which carry a truncating mutation on exon 21. Using the genetically-encoded calcium indicator GCaMP6f and two-photon calcium imaging, the authors found increased rate of calcium events at the dendritic spines of PNs in the prefrontal cortex of R1117X mutant mice. These events were greater in both frequency and amplitude compared to control animals and pointed to a hyperexcitability phenotype in PNs. But that’s only half the story…
Reduced dendritic inhibition by SST+ INs
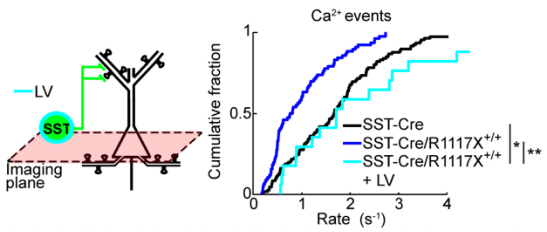
Previous literature4 suggests a significant role for somatostatin-positive (SST+) INs in regulating calcium signaling at dendritic spines. To test whether this may be a factor in the observed aberrant dendritic calcium transients, the authors generated double-transgenic R1117X mice which express GCaMP6s exclusively in SST+ INs. They observed a reduction in calcium events and reduced NMDAR signaling in mutant SST+ INs. This reduction in activity likely affects the overall control which INs are able to exert within the prefrontocortical microcircuitry and subsequently manifests as changes in the animal’s behavior.
Selective NMDAR overexpression in SST+ INs rescues behavioral deficits
The behavioral symptoms observed in R1117X mice include increased anxiety, reduced motor activity, and in the case of the prefrontal cortex, a severe deficit in associative learning. Electrophysiological data pointed to NMDAR as a possible target for rescue as they found reduced, but not eliminated, NMDAR currents. The authors elegantly parse out the phenotypes associated with IN dysfunction by selectively rescuing NMDAR currents in SST+ INs using lentiviral vectors to overexpress NMDAR subunits GluN1 and GluN2B. The overexpression of these in SST+ INs was sufficient to restore the rate of dendritic calcium events to a rate comparable to control animals. Additionally, this treatment rescued certain behavioral deficits, specifically trace fear learning and sensorimotor gating, suggesting these were mediated by reduced dendritic inhibition within the prefrontal cortex.
Made available under a CC-BY-NC-ND 4.0 International License.
Why this preprint?
I chose this preprint because of my general interest in IN development and their roles in disease. Studies indicate that ASD affects approximately 1:160 children worldwide, though numbers vary by country5. Government health agencies and NGOs spend significant resources on research to elucidate the underlying mechanisms of this group of heterogeneous disorders. It is through these efforts that numerous candidate genes have been implicated in ASD, but many studies to date place an emphasis on the effects of mutations in excitatory neurons. The authors of this study used robust assays to convincingly link the underlying interneuronopathy to behavioral symptoms found in mouse models of ASD. They achieved this by succinctly describing how altered inhibitory signaling contributes to increase in calcium signaling at the dendritic spines and ultimately in behavioral changes.
My questions for the authors:
- You mentioned Shank3B het mice had a decreased rate of calcium events (Fig. 2i). Do you have any clues as to which cell type would be primarily responsible for this? Is the interneuron hyperactive or is the pyramidal neuron less sensitive to stimuli? Maybe both?
- All of your experiments are done in adolescent mice and in the discussion you mention that calcium homeostasis during adolescent periods is critical for circuit maturation in the prefrontal cortex. A big question in the ASD field is whether or not certain deficits are reversible. Did you attempt any of the rescue experiments in older animals? At what point do the R1117X mutant mice stop responding to the GluN2B treatments?
References
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–267. doi:10.1034/j.1601-183x.2003.00037.x
- Žiburkus J, Cressman JR, Schiff SJ. Seizures as imbalanced up states: excitatory and inhibitory conductances during seizure-like events. J Neurophysiol. 2013;109(5):1296–1306. doi:10.1152/jn.00232.2012
- Dong Z, Chen W, Chen C, et al. CUL3 Deficiency Causes Social Deficits and Anxiety-like Behaviors by Impairing Excitation-Inhibition Balance through the Promotion of Cap-Dependent Translation. Neuron. 2020;105(3):475–490.e6. doi:10.1016/j.neuron.2019.10.035
- Marlin JJ, Carter AG. GABA-A receptor inhibition of local calcium signaling in spines and dendrites. J Neurosci. 2014;34(48):15898–15911. doi:10.1523/JNEUROSCI.0869-13.2014
- Elsabbagh M, Divan G, Koh YJ, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5(3):160–179. doi:10.1002/aur.239
doi: https://doi.org/10.1242/prelights.17014
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the neuroscience category:
Behavioral characteristics of an extremely old rhesus macaque in a zoo: Dementia-like symptoms and implications for quality of life of geriatric animals
Stefan Friedrich Wirth
EBV reprograms autoreactive anti-CNS B cells as antigen presenting cells in multiple sclerosis
Léa Bastien et al.
The Endocannabinoid System’s Contribution to Placebo Analgesia
Thomas Nicodemo Arrieta et al.
preLists in the neuroscience category:
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
SDB 78th Annual Meeting 2019
A curation of the preprints presented at the SDB meeting in Boston, July 26-30 2019. The preList will be updated throughout the duration of the meeting.
| List by | Alex Eve |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Young Embryologist Network Conference 2019
Preprints presented at the Young Embryologist Network 2019 conference, 13 May, The Francis Crick Institute, London
| List by | Alex Eve |