








Acorde: unraveling functionally-interpretable networks of isoform co-usage from single cell data
Posted on: 25 May 2021
Preprint posted on 9 May 2021
Arzalluz-Luque et al. present acorde, a computational pipeline that integrates bulk long read and single-cell short read RNA-seq to quantify isoform co-expression and co-usage networks at single-cell resolution.
Selected by Bobby RanjanCategories: bioinformatics, genomics
Background
There are a number of relevant questions regarding the importance of splicing for cell identity and function that can only be resolved by evaluating isoform expression at the single-cell level.
Integrating alternative splicing (AS) and gene expression changes has led to the discovery of cell subtypes and states that were otherwise not detected, thus hinting at the presence of coherent isoform variation. Further, co-expression relationships between transcript variants from different genes have not yet been investigated.
However, the uncertainty of short read-based isoform quantification and the heavy 3’ end bias of popular scRNA-seq methods has made investigating alternative splicing and isoform expression dynamics a challenging task. Although long read sequencing is able to alleviate these issues, its intrinsic sequencing depth constraints result in limited isoform diversity.
In this preprint, Arzalluz-Luque et al. present acorde – an end-to-end, data-intensive pipeline that integrates bulk long-reads and scRNA-seq to quantify and analyse isoform expression at single-cell resolution. They applied this pipeline to study the mouse primary visual cortex, using published scRNA-seq Smart-Seq2 (Tasic et al.) and bulk ENCODE PacBio long-read (Wyman et al.) data.
Key Findings

1. acorde provides an end-to-end computational pipeline to quantify and analyse isoform expression at single-cell resolution
acorde employs a hybrid strategy where bulk long-reads and single-cell short reads are integrated to estimate isoform expression at the single cell level. To alleviate the limitations of extant correlation metrics in the single-cell context, Arzalluz-Luque et al. developed a novel strategy to obtain noise-robust correlation estimates in scRNA-seq data, and a semiautomated clustering approach to detect modules of co-expressed isoforms across cell types (together known as the percentile correlation-based clustering approach). The authors additionally re-defined and implemented Differential Isoform Usage (DIU) and coDifferential Isoform Usage (coDIU) analyses in order to leverage the multiple cell types contained in single-cell datasets. Finally, they incorporated a functional annotation step in which several databases and prediction tools were integrated to add isoform-specific functional information
(Figure 1).
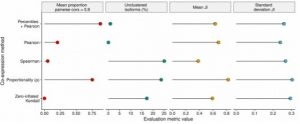
2. Percentile correlation-based clustering outperforms existing correlation and ρ proportionality metrics.
The percentile correlation-based clustering proposed as part of acorde was benchmarked against Pearson, Spearman and zero-inflated Kendall correlations, and the ρ proportionality metric. Of the 5 strategies compared, ρ proportionality came closest to the percentile correlation, but failed to control for unclustered transcripts (Figure 2).
3. Isoform selection exhibits cell-type-specific variation
To quantify the expression of the long read-defined isoforms at the single-cell level, the authors applied acorde to study the mouse primary visual cortex using published scRNA-seq Smart-Seq2 (Tasic et al.) and bulk ENCODE PacBio long-read (Wyman et al.) data. Interestingly, the number of coDIU genes linking isoform co-expression clusters was dependent on cluster sizes, but showed no direct relationship with the similarities between expression profiles, suggesting that coordinated isoform usage mechanisms may produce strong cell type-level shifts in isoform selection. Indeed, in the Tasic et al. dataset, a high proportion of coDIU interactions were detected for highly expressed isoforms in neural cell types. While isoform clusters with high neuronal expression were among the largest in size, it may be plausible that co-splicing be at the core of primary visual cortical neural function regulation.
4. Isoform co-expression may be post-transcriptionally regulated
Annotating the genes regulated by coDIU revealed a specific enrichment of mitochondrial components, suggesting that coordinated isoform usage may affect oxidation and energy
metabolism. Interestingly, coDIU genes also showed additional enrichment for splicing-related terms such as RNA splicing, mRNA splicing via spliceosome and for 3’ UTR motif K-box. This result links genes involved in splicing and RNA stability with the coordination of AS, and suggests that co- expression of alternative isoforms is a post-transcriptionally regulated process.
5. coDIU genes demonstrate potential cell-type-specific splicing-mediated functional synergy
The authors then focused on coDIU genes representing 3 clusters of isoforms: oligodendrocyte- specific, neuron-specific and shared isoform expression patterns.
They found that the K-box motif, which has been proposed as a negative post-transcriptional regulator, presented inclusion changes in ~60% of annotated coDIU genes. In addition, the coDIU network included several genes in which 3’UTR elongation led to neuron-specific co-inclusion of K- box motifs, some of which may be involved in neuron survival and differentiation. This suggests a 3’ UTR binding-mediated mechanism favouring isoform co-expression may regulate post- transcriptional modifications of neuron survival genes.
The majority of neuron-oligodendrocyte coDIU genes also presented coding region variation – protein domains (PFAM) and post-translational modifications (PTMs). Two tubulin isotypes, Tubg2 and Tubb4b, had co-expressed isoforms with neuron-specific and neuron-oligodendrocyte expression, respectively. Both genes presented inclusion changes in an N-terminal GTP-ase domain and several PTMs with differing functional outcomes, suggesting a cell-type-specific fine-tuning mechanism for modifying tubulin stability and its interactions with other proteins (the “tubulin code”).
Why I chose to highlight this preprint
This preprint tackles an important challenge in the single-cell field – the analysis of isoform variation and co-expression at single-cell resolution. acorde provides a robust solution to quantify isoform variation by mapping on to reference long reads. The percentile correlation-based approach provides a novel solution for tackling the noise in scRNA-seq correlations. This preprint demonstrates the relevance and capabilities of acorde in the analysis of isoform co-usage at single- cell resolution.
Questions for the authors
- Has acorde been tested on 10X Genomics 5’ or 3’ scRNA-seq data? How does its performance compare to Smart-Seq2 data?
- What is the false-positive rate for percentile correlations, and does it suffer from spurious correlations as compared to traditional approaches?
- Can acorde be used to perform a case-control analysis to study disease-specific isoform variation?
doi: https://doi.org/10.1242/prelights.29126
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |