








Brain deletion of Serf2 shifts amyloid conformation in a mouse model
Posted on: 6 April 2021 , updated on: 15 April 2021
Preprint posted on 3 March 2021
Serving as an aggregation modifier: SERF2 shifts the amyloid aggregation process in mammalian brain
Selected by Utrecht Protein Folding and AssemblyCategories: molecular biology
PreLight written by Ruben van Diest, Myrthe Franken, Sanne Pikaar and Nian Vervoort
Background
Neurodegenerative diseases, such as Alzheimer’s disease, are characterized by protein aggregation in the brain. In Alzheimer’s disease, the protein amyloid-β (Aβ) clumps together into fibrils and forms amyloid plaques outside the cells. Recent research with brain samples of diseased patients has demonstrated that variants of amyloid fibrils exist, even when formed by the same protein [1]. Structural variants of Aβ fibrils are suspected to affect aggregate toxicity and disease progression [1]. In 2010, a modifier of the aggregation process (MOAG-4) was discovered in C. elegans, which has SERF1a and SERF2 as its human orthologs [2]. While their function remains largely unknown, MOAG-4 and SERF were able to stimulate aggregation and enhance toxicity in C. elegans and human cell lines, respectively [2]. A structural study has revealed that the binding of SERF1a causes a conformational change in an aggregation-prone protein, increasing its aggregation tendency [3]. These results led to the hypothesis that MOAG-4/SERF affects the aggregation process. The same authors who discovered MOAG-4/SERF now explore the effects of a Serf2 knock-out (KO) on the aggregation process of Aβ in a more complex mammalian system, the mouse.
Key results
The authors first set out to generate a mouse model to study the effects of SERF2. Full-body Serf2 KO mice are however not viable due to severe developmental issues, suggesting a role for SERF2 in early development. Instead, the authors develop brain-specific Serf2 KO (Serf2br-/-) mice which are viable, and cross these with amyloid model (AM) mice. In this AM model the Alzheimer amyloid precursor protein (APP) with disease mutations is overexpressed. The authors use the resulting AM and AM;Serf2br-/- (here referred to as AM and Serf2 KO, respectively) for further experiments. They confirm that the levels of the APP cleavage products, Aβ40 and Aβ42, in these mice are unaffected by the brain-specific knock-out of Serf2.
The authors then investigate the effects of SERF2 elimination on the aggregation process by staining the Aβ aggregates in brain samples from Serf2 KO mice and AM mice with antibodies. They do not identify distinct differences in plaque morphology between the Serf2 KO mice and the AM mice. Quantification of the stained plaques shows a correlation between Aβ levels and the number of plaques. Surprisingly, the authors observe a slight increase in plaque load for the Serf2 KO mice compared to the AM mice with similar Aβ levels. They also detect a higher amount of intracellular staining in the Serf2 KO mice relative to the AM mice, suggesting that the absence of SERF2 influences Aβ aggregation already inside the cell before formation of extracellular plaques.
Next, the authors stain the brain samples with Thioflavin-S (ThS), a compound that binds amyloid fibrils. Although they do not observe a difference in ThS plaque load between the Serf2 KO and the AM groups, a distinction between the groups is noticeable when they compare the number of ThS plaques with the Aβ levels in individual animals. The authors find a positive correlation between the Aβ levels and the number of ThS-positive plaques in the AM mice, while this correlation is negative for the Serf2 KO mice, indicating that a loss of SERF2 induces a change in aggregation behaviour.
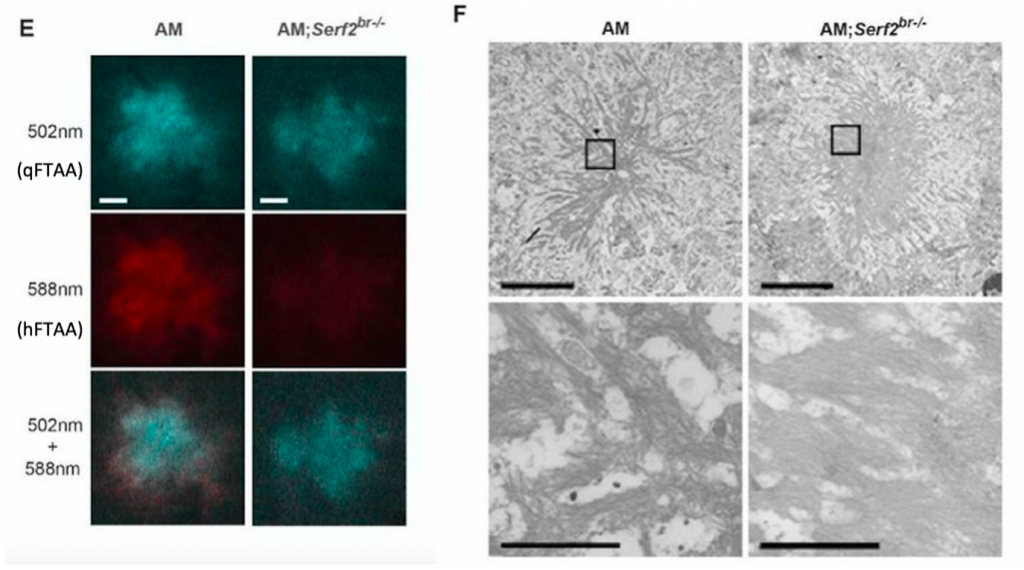
The authors follow up on this result using two luminescent amyloid binding dyes. The first dye, qFTAA, only binds to mature amyloids, just like ThS. The other dye, hFTAA, binds to mature amyloids as well as to the less densely packed immature ones. With spectroscopy they measure the ratio between the two dyes in individual plaques. They discover that the plaques in the Serf2 KO have on average a higher qFTAA:hFTAA ratio compared to the plaques in the AM mice. Light microscopy shows that qFTAA binds the plaques of both groups in the same manner. However, the plaques of the AM mice contain hFTAA in the core and the border, while hFTAA staining is hardly present in the plaques of the knockout mice (Figure 4E). This indicates that the plaques of Serf2 KO mice contain fewer immature amyloids and are generally more compact.
With scanning transmission electron microscopy (STEM) the authors visualize the global fibril morphology in greater detail. The images reveal that the Serf2 KO plaques are indeed more condensed compared to those of the AM mice (Figure 4F). With this, the authors conclude that Serf2 KO causes a denser plaque morphology in mice, indicating a conformational shift by SERF2 in the Aβ aggregation process.
Why we chose this preprint
As life sciences bachelor students, we have been educated on the topics of protein folding, aggregation and neurodegenerative diseases. We are fascinated by the intricacy of the aggregation process and how modern science tries to understand and cure diseases such as Alzheimer’s disease. We are excited about this preprint because it provides a new perspective in the aggregation process of specifically Aβ, but likely also other aggregation-prone proteins. By confirming the ability of the endogenous factor SERF2 to alter the aggregation process in their mouse model, the authors are the first to describe the action of an aggregation modifier in a more complex organism. These results might provide the basis for the identification of other endogenous factors that influence aggregation and might explain why there are structural variants of Aβ plaques in Alzheimer patients. We are curious to see what these results will mean for future therapeutic strategies.
Questions
In your studies, you explore the effects of MOAG-4/SERF in both C. elegans and mouse models. In C. elegans you describe a decrease in aggregation upon deletion of moag-4, while you do not observe this decrease in the mouse models. How would you interpret these results?
For this study you have put a lot of effort into creating a brain-specific KO mouse model. How can your model contribute more to future studies in the field?
Do you the think the differences you found between the amyloid plaques could affect the disease progression of Alzheimer in the mice? Do you have ideas how to further investigate this?
How would you imagine your results to contribute to finding a treatment for neurodegenerative diseases?
References
[1] Qiang, W., Yau, W.-M., Lu, J.-X., Collinge, J. & Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 541, 217–221 (2017)
[2] van Ham, T. J. et al. Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612 (2010)
[3] Merle, D. A. et al. Increased Aggregation Tendency of Alpha-Synuclein in a Fully Disordered Protein Complex. J. Mol. Biol. 431, 2581–2598 (2019)
doi: Pending
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the molecular biology category:
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Defective BRCA1-mediated DNA end resection drives tandem duplication formation and FANCM synthetic lethality
Marta San Martin
preLists in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |