








Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain (AL) amyloidosis patient
Posted on: 5 November 2018 , updated on: 20 November 2018
Preprint posted on 17 October 2018
Article now published in Nature Communications at http://dx.doi.org/10.1038/s41467-019-09133-w
Zooming in on protein misfolding diseases - the first high-resolution structure of patient-derived light chain amyloid fibrils revealed
Selected by Tessa SinnigeCategories: biochemistry
Background
A large number of proteins that are unrelated in sequence or structure are prone to misfold into amyloid fibrils, which have a characteristic cross-β structure and are associated with a wide variety of human diseases1. Perhaps best known are the proteins that misfold in the brain, e.g. amyloid-β and tau in Alzheimer’s disease, and α-synuclein in Parkinson’s disease. However, protein fibrils can be deposited in various other organs, giving rise to systemic amyloidosis. Amyloid light chain (AL) amyloidosis is the most common of these diseases, and is caused by the clonal expansion of a plasma cell that overproduces an immunoglobulin light chain, which forms fibrils upon secretion into the extracellular space. The amyloid deposits can accumulate in different organs, with their presence in the heart giving the worst prognosis.
For a long time it has been difficult to solve high-resolution structures of amyloid fibrils given that they are generally not crystalline, but this has changed with the recent advances in cryo-electron microscopy (cryo-EM). A noticeable break-through in the field of protein misfolding diseases is that we can now solve atomic resolution structures of patient-derived amyloid fibrils, as was first shown in the case of tau fibrils extracted from Alzheimer’s disease brain2. Although the precise relationship between amyloid-like protein misfolding and cellular toxicity remains to be established, these structures are expected to contribute to a better understanding of the disease process and inspire drug design.
The results
In this preprint, the first high-resolution structure of amyloid fibrils from a patient with systemic amyloidosis is reported. Light chain fibrils were isolated from heart tissue of an AL patient, and the cryo-EM structure was solved at 4.0 Åresolution. First however, the composition of the fibrils was assessed, and the authors identified a mixture of different lengths of the light chain, ranging from the full-length protein to just the variable domain, VL. Limited proteolysis showed that the VLdomain was protected against degradation, suggesting that it formed the fibril core, whereas the C-terminal parts were efficiently degraded by the protease.
A previous study on patient-derived light chain fibrils at lower resolution showed that the fibrils were polymorphic3, i.e. existing in distinct structural forms, but in this case only one type of structure was identified on the EM grids. The cryo-EM map revealed a density which at first sight looks like a symmetric dimer of protofibrils. However, the authors show that this is not the case, and in fact the fibril is built up from asymmetric layers composed of a single protein monomer. Two regions of the VL domain could be fitted in the density as curved β-strand segments, leaving a large part of less defined density open. This could potentially arise from disordered protein segments surrounding the fibril core, similar to the “fuzzy coat” around the core of the brain-derived tau fibrils2. However, given that the VL domain was resistant to proteolytic digestion, the region connecting the two fitted segments is likely to be ordered, yet heterogeneous in conformation. The other, larger portion of density that could not be fitted might be occupied by C-terminal extensions of the protein, corresponding to the longer protein fragments that were shown to be present in the fibrils in addition to the VL domain only.
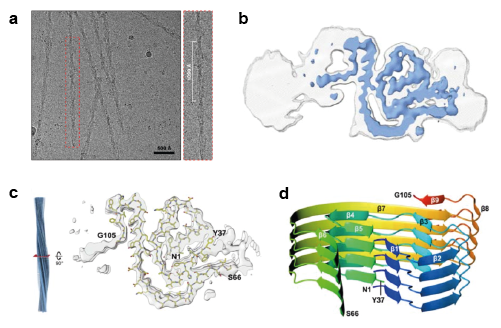
a) Light chain fibrils extracted from the AL patient’s heart tissue on the EM grid. b) Cross-sectional cryo-EM map sharpened at 4.0 Å (blue) and unsharpened at 4.5 Å (grey). c) Part of the VL sequence modelled into the density. d) Ribbon representation of four stacked layers of the fibril core showing the β-strand segments. Adapted from the preprint under a CC-BY 4.0 International license.
One of the key questions in the field of AL amyloidosis is how the light chain sequence correlates with its aggregation propensity. Normally, a library of light chain variants that can recognise specific antigens is created by genetic shuffling of the domains followed by somatic hypermutation. In AL amyloidosis patients, however, one particular light chain variant is overproduced by monoclonal plasma cells, which has raised the question if these sequences are more prone to amyloid formation than regular light chains. At the same time, one might wonder how the different sequences from individual patients could accommodate a similar fibril fold. The structure reported here shows that some of the particularly variable regions are embedded in the fibril core and contribute to the β-strand segments, but the residues that are critical for the structural contacts appeared largely conserved when a panel of disease-associated light chain sequences was compared. Light chains from different AL patients could thus form amyloid fibrils with similar characteristics as the structure here described, although it remains to be determined whether sequence-related changes in morphology may occur.
Why I chose this preprint
I have always been fascinated by amyloid fibrils, as they appear to represent a universal fold that virtually any polypeptide can adopt, and they are a hallmark of a large number of diseases although the mechanisms linking protein aggregation to toxicity are still poorly understood. Also, these fibrils are such beautiful structures! The first structural models of fibrils formed by short peptides and yeast prion proteins appeared during my studies in biochemistry, and really sparked my interest in the field of protein misfolding. It is very exciting to see that the revolution in cryo-EM now allows scientists to solve fibril structures directly from human material. After recent work on brain-derived tau fibrils2,4,5, this preprint shows the first high-resolution structure of fibrils from an amyloidosis patient. I find the large region of density that could not be modelled somewhat unsatisfying, but at the same time it is a good example of the fact that amyloid fibrils are not necessarily uniform and rigid, but may have dynamic or heterogeneous regions protruding from their core, which could be important with respect to cellular interactions and toxicity.
Questions and outlook
The authors suggest that some of the less-defined density could be occupied by the part of the VL domain connecting the two modelled regions, whereas the other, larger region could arise from C-terminal extensions. I am wondering if the authors consider it worthwhile to confirm this by obtaining the cryo-EM map of the fibrils after limited proteolysis, which they showed removes the C-terminal parts but leaves the VL domain intact. Furthermore, can the authors speculate whether the C-terminal domain may retain its native fold in the context of the fibrils, or C-terminal regions may participate in fibril formation by adopting a cross-β structure? If they were completely unfolded and extended away from the core, they would probably not give rise to any density in the cryo-EM map.
Finally, I am looking forward to more fibril structures being solved from AL patients with different light chain variants, which will be necessary to better understand the effect of the amino acid sequence on fibril structure and polymorphism.
References
- Chiti, F. & Dobson, C. M. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68 (2017).
- Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
- Annamalai, K. et al. Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew. Chemie – Int. Ed. 56, 7510–7514 (2017).
- Falcon, B. et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140 (2018).
- Falcon, B. et al. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 136, 699–708 (2018).
doi: https://doi.org/10.1242/prelights.5384
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the biochemistry category:
Site-Specific Inhibition of Translation Initiation via 2’-O-methylation
Leonie Brüne
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the biochemistry category:
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
Peer Review in Biomedical Sciences
Communication of scientific knowledge has changed dramatically in recent decades and the public perception of scientific discoveries depends on the peer review process of articles published in scientific journals. Preprints are key vehicles for the dissemination of scientific discoveries, but they are still not properly recognized by the scientific community since peer review is very limited. On the other hand, peer review is very heterogeneous and a fundamental aspect to improve it is to train young scientists on how to think critically and how to evaluate scientific knowledge in a professional way. Thus, this course aims to: i) train students on how to perform peer review of scientific manuscripts in a professional manner; ii) develop students' critical thinking; iii) contribute to the appreciation of preprints as important vehicles for the dissemination of scientific knowledge without restrictions; iv) contribute to the development of students' curricula, as their opinions will be published and indexed on the preLights platform. The evaluations will be based on qualitative analyses of the oral presentations of preprints in the field of biomedical sciences deposited in the bioRxiv server, of the critical reports written by the students, as well as of the participation of the students during the preprints discussions.
| List by | Marcus Oliveira et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Cellular metabolism
A curated list of preprints related to cellular metabolism at Biorxiv by Pablo Ranea Robles from the Prelights community. Special interest on lipid metabolism, peroxisomes and mitochondria.
| List by | Pablo Ranea Robles |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |