








SARS-CoV-2 Variants are Selecting for Spike Protein Mutations that Increase Protein Stability
Posted on: 4 August 2021 , updated on: 6 August 2021
Preprint posted on 25 June 2021
Article now published in Journal of Chemical Information and Modeling at http://dx.doi.org/10.1021/acs.jcim.1c00990
Categories: bioinformatics, biophysics
Highlighted by
Soni Mohapatra1 and Alexander Solis*, 1
*Undergraduate Researcher, (1) Johns Hopkins University, Baltimore, USA
Background
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused huge disruption across the globe, resulting in over 4 million deaths as of August 2021 (1). The highly contagious nature of SARS-CoV-2 can be attributed to its molecular structure. In particular, the structural spike proteins which radiate from the protein center bind with a high affinity to ACE2 (human angiotensin-converting enzyme-2), which acts as a receptor for the virus and is expressed in a variety of human organs (2). Errors during RNA replication of the virus as well as evolutionary pressures has led to the rise of mutant strains as the pandemic spreads. Mutations in spike proteins could lead to increased ACE2 receptor binding, changes to cleavage/glycosylation sites (3), increased protein stability, and/or better infectivity. A thorough understanding of the mutational landscape of the spike proteins, as well as the ability to predict deadly mutations, could therefore be paramount for vaccine and therapeutic drug production, as well as to inform public health measures.
Changes in Gibbs free energy of unfolding (ΔΔG) between the mutant and the original protein, is a thermodynamic parameter for predicting how mutations in a protein affect the stability of the protein. ΔΔG = ΔGmutant − ΔGwild‑type, where ΔG is the difference between the free energies of the folded and unfolded states. Typically, ΔΔG < 0 indicates that the mutations are stabilizing whereas a ΔΔG > 0 suggests that mutations are destabilizing. As such, it can be used to obtain insights regarding possible future mutations and their effects on the protein as a whole. Mutations of SARS-CoV-2 spike proteins that increase protein stability could lead to the survival of the protein in different environmental conditions and therefore result in a longer lifespan of the protein.
In this preprint, the authors calculated the respective ΔΔG for all possible missense mutations for the SARS-CoV-2 spike protein as a means of understanding how spike protein stability has and will evolve over time.
Key findings
Evolutionary enrichment of mutations that stabilize spike proteins
The authors calculated the ΔΔG for all possible 19440 missense mutations of the spike protein using the most recently published structure (PDBID: 6XVV). Mutations with ΔΔG < -1 kcal/mol and ΔΔG > 2.5 kcal/mol were defined as strongly stabilizing and strongly destabilizing mutants, respectively. Only 3.9% of these ‘background’ missense mutations were found to be strongly stabilizing. They compared these ‘background’ mutations to those that are found in the WHO “Variants of Concern” (VOC) and “Variants of Interest” (VOI) categories. Interestingly, ΔΔG of mutations occurring in the above two categories are significantly lower than the overall ΔΔG of the ‘background’ mutations. 4 of the 17 mutations observed in the VOC had a ΔΔG <= ~ -1 kcal/mol. Additionally, it was noted that none of the mutations present in the VOC category were strongly destabilizing. In stark contrast, 34% of the ‘background’ mutations are strongly destabilizing. Taken together, their data suggested that stabilizing mutations in the spike proteins are enriched in the WHO variants category compared to all ‘background’ missense mutations.
They also calculated the ΔΔG distribution of all the different mutations present in 10 variants belonging to VOC and VOI categories. 7 out of the 10 variants had a mean ΔΔG distribution below that of the mutational background (Fig. 1), which provided further support to the claim that stabilizing mutations are enriched in these categories compared to mutational background.
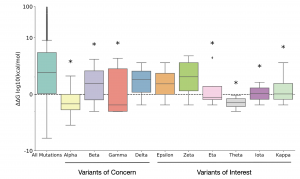
Fig. 1: Distribution of ΔΔG for mutations observed in the WHO VOC and VOI categories vs. background mutations (Taken from Fig. 3 of the preprint, made available by a CC-BY 4.0 license)
Predicting the potential evolutionary order of variants and gaining foresight into traits of future variants
The authors also used this calculation of ΔΔG to gather insight into the evolutionary order in which the mutations appeared in the variants. They calculated the ΔΔG for every possible combination of mutations in the different variants. For the Alpha variant, the gradual addition of mutations to the original spike protein resulted in lowering of ΔΔG and therefore increased stabilization of the variant compared to the original protein. In contrast, the resulting ΔΔG from the combination of the mutations in the Beta variant is destabilizing. The authors noted that the calculated ΔΔG is however still lower than that derived from a combination of mutations that could lead to the most destabilizing variant.
This analysis provided insight into evolutionary mechanisms for selective mutations that are employed by the variants. In their analysis, the authors find that mutations which lead to a more stable variant are generally favored over others. However, it was also alluded to that some combinations of mutations, which include those that are destabilizing, may ultimately create an evolutionarily superior product than the stabilizing mutations This is due to the fact that mutations have varying effects when expressed and can provide advantageous qualities, such as increased affinity to binding sites, better survival in different environmental conditions, amongst others. They therefore conclude that variant evolution does not only favor stabilizing mutations, but also those which carry properties that can positively manipulate a protein’s function.
Why we like the preprint
In this article, the authors make use of an accessible and easy-to-understand concept of ΔΔG as a means of comprehending the complex mechanics of protein stability in SARS-CoV-2. This was done through performing calculations of ΔΔG to measure the stability of present variants as well as using ΔΔG to understand the evolutionary order of different mutations in the variants. What we particularly enjoyed about the article is that the calculation of a simple biophysical parameter, ΔΔG makes understanding the evolution of SARS-CoV-2 accessible to a wider audience, but also allows one to predict the uprising of future deadly variants.
Questions for authors
- Is it possible to use the ΔΔG calculations to guide vaccine development?
- Are there mutations in the ‘background’ mutational scan that have a ΔΔG lower than the mutations occurring in the WHO categories? Are those the mutations we should watch out for in the future?
References
- “WHO Coronavirus (COVID-19) Dashboard.” World Health Organization, World Health Organization, 16 July 2021, covid19.who.int/.
- Zheng, Jun. “SARS-CoV-2: an Emerging Coronavirus that Causes a Global Threat.” International journal of biological sciences vol. 16,10 1678-1685. 15 Mar. 2020, doi:10.7150/ijbs.45053
- Tortorici, M. A.; Veesler, D. Structural Insights into Coronavirus Entry. In Advances in Virus Research; 2019; Vol. 105, pp 93–116. ISSN 0065-3527. ISBN 9780128184561. https://doi.org/10.1016/bs.aivir.2019.08.002.
doi: https://doi.org/10.1242/prelights.30208
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the biophysics category:
Mechanically-induced Septin Networks Protect Nuclear Integrity
Filipe Nunes Vicente
Loss of Sun2 ablates nuclear mechanosensing-driven extracellular matrix production and mitigates lung fibrosis
Beth Chopak
Shape independent fluidisation in epithelial monolayers
Sindhu Muthukrishnan
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the biophysics category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
66th Biophysical Society Annual Meeting, 2022
Preprints presented at the 66th BPS Annual Meeting, Feb 19 - 23, 2022 (The below list is not exhaustive and the preprints are listed in no particular order.)
| List by | Soni Mohapatra |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Biophysical Society Meeting 2020
Some preprints presented at the Biophysical Society Meeting 2020 in San Diego, USA.
| List by | Tessa Sinnige |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Biomolecular NMR
Preprints related to the application and development of biomolecular NMR spectroscopy
| List by | Reid Alderson |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |