








State-of-the-Art Estimation of Protein Model Accuracy using AlphaFold
Posted on: 13 April 2022 , updated on: 9 August 2023
Preprint posted on 24 March 2022
Article now published in Physical Review Letters at http://dx.doi.org/10.1103/PhysRevLett.129.238101
AlphaFold may not be “just” a pattern recognition algorithm, but may actually have learnt about the energetics of protein folding.
Selected by Kieran DidiCategories: bioinformatics, biophysics
Background
The field of protein structure prediction was revolutionized last year when the DeepMind team, which won the 14th Critical Assessment of Structure Prediction (CASP14) competition, published the paper2 and the code for their AlphaFold (AF2) model. This major advance on the protein folding problem holds the promise for progress in many areas of biology and medicine, since protein structures are essential for research in e.g. drug discovery and protein engineering, but often only accessible via expensive and laborious experimental methods such as X-ray crystallography, cryo-electron microscopy and NMR spectroscopy. These methods have enabled scientists to elucidate more than 100,000 protein structures (available through the Protein Data Bank), but are costly and involve a lot of trial and error. Computational methods try to simplify this procedure by predicting the 3D structure of a linear protein sequence without experimentally determining it.
Early efforts at simplifying protein structure prediction aimed to capture the physics that govern protein folding and simulate the folding process to get an accurate structure; a prime example of this is the Rosetta software suite developed by David Baker and co-workers at the University of Washington in Seattle. In the 90s, coevolution information was recognized as a valuable input for protein structure prediction. For this, multiple sequence alignments (MSAs) between evolutionarily related proteins are constructed and spatial contacts are inferred based on coevolution of amino acids. Finally, the progress in the field of machine learning and especially deep learning also had an impact on the structural biology community, with huge models such as AlphaFold producing state-of-the-art protein structure predictions.
One open question regarding AlphaFold is whether the model learned something about the underlying physics of the protein folding problem or is “just” a pattern matching algorithm inherently dependent on the provided MSAs. Since proteins in nature fold astonishingly fast by themselves (a phenomenon known as Levinthal’s paradox) and some of them can refold after denaturation (as observed by Anfinsen3), the 3D structure must be encoded in the protein sequence alone. Anfinsen’s dogma, therefore, states that proteins fold as a result of free energy minimization. This free energy depends on the protein structure and is the one that earlier physics-based prediction tools tried to approximate (and that is, for example, still approximated in techniques like molecular dynamics simulations).
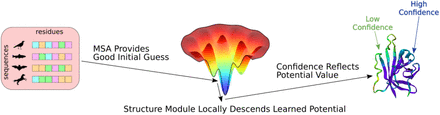
In this preprint, Roney and Ovchinnikov address this question by testing the hypothesis that AlphaFold learned this energy function and uses coevolution information to find a good initial guess for an energy minimum in this conformation landscape, therefore understanding something about the underlying physics of the protein folding problem. They use this hypothesis to rewire AlphaFold in such a way that they can rank decoy protein structures with it, performing better than state-of-the-art (SOTA) models for this task.
Key findings
Use of AF2 for ranking candidate protein structures
During the structure prediction process, AlphaFold uses an MSA of the amino acid sequence of the target protein with related sequences as input. As an additional option, known protein structures close to the target protein sequence (known as templates) can be provided to improve prediction results. The model then outputs a predicted protein structure and two confidence metrics for this prediction: the predicted LDDT-Cα Score (pLDDT) and the predicted TM Score (pTM).
To change the objective of AlphaFold from predicting protein structures to ranking candidate structures, the authors made three adjustments. First, instead of providing known protein structures as templates, they provide a “decoy structure” that is a candidate structure for the target protein, e.g. one predicted by another model. Furthermore, they do not provide an MSA as input, but just the amino acid of the target protein, therefore stripping the model of the ability to use coevolutionary information. Lastly, they compute a new output metric called a “composite confidence score” based on the existing metrics: they multiply output pLDDT, output pTM and the TM Score between the structure predicted by AlphaFold and the decoy. The last term is needed since the main objective is not to assess the quality of the predicted structure, but the quality of the decoy structure that was given as a template.
The authors use this approach to rank decoys from the Rosetta decoy dataset, which contains 133 native protein structures along with thousands of decoy structure variants, and compare the performance with common decoy ranking tools such as Rosetta4 and the SOTA machine learning model DeepAccNet5. Their approach based on AlphaFold strongly outperforms Rosetta and DeepAccNet, both in terms of Spearman correlation of the confidence metric with decoy quality and in terms of top-1 accuracies of decoy structures.
Ranking quality independent of decoy’s amino acid sequence
The decoy structures the authors provide the model with masks out the side chains that help to increase the accuracy. Since the decoy structure now basically consists of the backbone and the Cβ-atoms only, any sequence of correct length could be fed into the model as input instead of the correct sequence of the target protein. The authors investigated the influence of this parameter by running their experiments with two different one-hot-encoded sequence inputs: both the true target sequence and an all-alanine sequence. They found that both choices deliver robust results on the Rosetta decoy dataset, with the all-alanine sequence performing better on the correlation metric and the correct target sequence performing better on the top-1 accuracies. The authors then used this result to further extend their hypothesis regarding the inner workings of their decoy ranking predictions: in the case of the target sequence input, this sequence and the masked-out sequence of the template are identical, and therefore the structural predictions probably very similar. Since the global geometry is thus quite similar, the confidence metrics used to compute the composite score are therefore more dependent on local fold features, delivering better results on the top-1 accuracies.
For the all-alanine sequence, the opposite is the case: due to very low sequence similarity, the global symmetry between decoy and prediction will be very different, causing the confidence metrics to be strongly influenced by the global fold and the model to perform better on the general correlation metric. Using a weighted hybrid approach, the authors were able to combine the strengths of both methods and outperform the results gained by providing either of the inputs alone.
Evaluation from CASP14: MSAs needed for accurate structure prediction, not for decoy ranking
To test their hypothesis on an independent data set, the authors used the CASP14 EMA (Estimation of Model Accuracy) tasks. Here, they show that AlphaFold is indeed able to rank decoys better than the top models from CASP14 without coevolution information, but still needs the MSAs to perform structure prediction itself. Without MSAs, it can rank predicted decoys reliably but performs poorly in producing structure predictions, further supporting the author’s hypothesis that coevolution information is used to provide a good initial guess on the learned energy landscape, from which the structure module performs local gradient descent to an energetic minimum.
Why I selected this preprint
The publication of AlphaFold had a major influence on the structural biology community and the life sciences in general by improving experimental structure predictions, as well as providing thousands of predicted structures to researchers around the world. However, more difficult problems such as protein design still pose a challenge. The main hypothesis of this preprint (i.e. that AlphaFold has learnt some kind of underlying energy function) presents a novel idea that suggests new angles from which challenging problems in structural biology can be tackled.
Questions for the authors
1. The preprint provides evidence for your hypothesis that AF2 learns an energy function for protein folding, but what other experiments could be used to support/falsify your hypothesis?
2. For protein structure prediction, the MSAs still seem indispensable. If your hypothesis is true, in what ways could this new insight be used for problems such as protein design/structure prediction for single sequences?
References
(1) Roney, J. P.; Ovchinnikov, S. State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold. bioRxiv March 12, 2022, p 2022.03.11.484043. https://doi.org/10.1101/2022.03.11.484043.
(2) Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S. A. A.; Ballard, A. J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A. W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2.
(3) Anfinsen, C. B.; Scheraga, H. A. Experimental and Theoretical Aspects of Protein Folding. In Advances in Protein Chemistry; Anfinsen, C. B., Edsall, J. T., Richards, F. M., Eds.; Academic Press, 1975; Vol. 29, pp 205–300. https://doi.org/10.1016/S0065-3233(08)60413-1.
(4) Rubenstein, A. B.; Blacklock, K.; Nguyen, H.; Case, D. A.; Khare, S. D. Systematic Comparison of Amber and Rosetta Energy Functions for Protein Structure Evaluation. J. Chem. Theory Comput. 2018, 14 (11), 6015–6025. https://doi.org/10.1021/acs.jctc.8b00303.
(5) Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved Protein Structure Refinement Guided by Deep Learning Based Accuracy Estimation. Nat. Commun. 2021, 12 (1), 1340. https://doi.org/10.1038/s41467-021-21511-x.
doi: https://doi.org/10.1242/prelights.31785
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
A potential anti-amyloidogenic therapy for type 2 diabetes based on the QBP1 peptide
Joao Gabriel, Marcus Oliveira
The lipidomic architecture of the mouse brain
CRM UoE Journal Club et al.
Kosmos: An AI Scientist for Autonomous Discovery
Roberto Amadio et al.
Also in the biophysics category:
Liquid-Liquid Phase Separation-mediated formation of amyloid fibrils from DcpS scavenger enzymes
Joao Gabriel, Marcus Oliveira
A potential anti-amyloidogenic therapy for type 2 diabetes based on the QBP1 peptide
Joao Gabriel, Marcus Oliveira
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the biophysics category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
66th Biophysical Society Annual Meeting, 2022
Preprints presented at the 66th BPS Annual Meeting, Feb 19 - 23, 2022 (The below list is not exhaustive and the preprints are listed in no particular order.)
| List by | Soni Mohapatra |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Biophysical Society Meeting 2020
Some preprints presented at the Biophysical Society Meeting 2020 in San Diego, USA.
| List by | Tessa Sinnige |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Biomolecular NMR
Preprints related to the application and development of biomolecular NMR spectroscopy
| List by | Reid Alderson |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |