








Structure of an infectious mammalian prion
Posted on: 4 March 2021
Preprint posted on 15 February 2021
Article now published in Molecular Cell at http://dx.doi.org/10.1016/j.molcel.2021.08.011
Categories: biochemistry
Background:
Neurodegenerative diseases are highly debilitating conditions. Collectively, they cause a gradual degeneration of the central or peripheral nervous system, with symptoms ranging from memory loss, mood swings and behavioural changes to a loss of movement (depending upon the locations of the affected nerves; 1). What triggers neurodegeneration is still widely investigated however a group of such disorders (for example Creutzfeldt-Jakob Disease; CJD) have been linked to lethal prion pathogens. Prions are very unusual pathogens as, unlike viruses, bacteria and parasites, they do not contain DNA or RNA. Instead, prions are infectious, misfolded assemblies of a normal cellular prion protein (PrPC), with the resulting protein known as PrPSC. Prions can self-propagate, meaning PrP monomers assemble as fibrils, which can fragment then initiate the formation of other fibrils (2). They transmit in various ways, for example via blood transfusion and infected tissue/other fluids, consumption of infected tissues or from the environment from contaminated soil (3). The pathology of the infection arises when the prion form of the protein forms aggregated (often in fibrillar forms called amyloids) which accumulate and fatally damage tissue; a similar pathology can be observed in diseases such as Parkinson’s and Alzheimer’s disease (4). As development of a prion disease is centred on the way in which the PrPC folds in the host organism, understanding how prions fold and multiply is key to understanding disease formation, transmission and manifestation. Here, the authors perform Cryo-EM to produce the first high-resolution structure of an infectious mammalian prion. This prion strain (named 263K) was purified from the brains of terminally ill hamsters.
Key Findings:
PrPC is a cell surface protein attached to the surface via a glycosylphosphatidylinositol (GPI) anchor. The N-terminal section is disordered and flexible whereas the C-terminal is made up of 3 alpha-helical domains, and a small beta-sheet. PrPC is both protease sensitive (i.e can be digested) and detergent soluble which is entirely contrary to the prion form (PrPSC) of the protein which is highly resistant to both. PrPSC assembles as beta-sheet rich fibres, that can appear like twisted ribbons or intertwined filaments. This arrangement is starkly different to the arrangement of the original non-prion form of the protein which is largely alpha helical with a very low beta-sheet content (5). In this study, the authors use cryogenic electron microscopy (Cryo-EM) to produce a structure of the prion form of the protein. Cryo-EM is a sophisticated, high resolution microscopy technique in which samples can be imaged in their native hydrated state. Samples are ‘flash frozen’ in a cryogenic (extremely low temperature) solution, for example, liquid ethane. This super rapid freezing means water molecules within the sample will be vitrified rather than crystallized, instead leaving a solid sample in its native hydrated state which can be imaged at high resolution on a transmission electron microscope (which uses a high-energy particle beam to capture the pattern of electron scatter generated when the beam passes through the sample; 6).
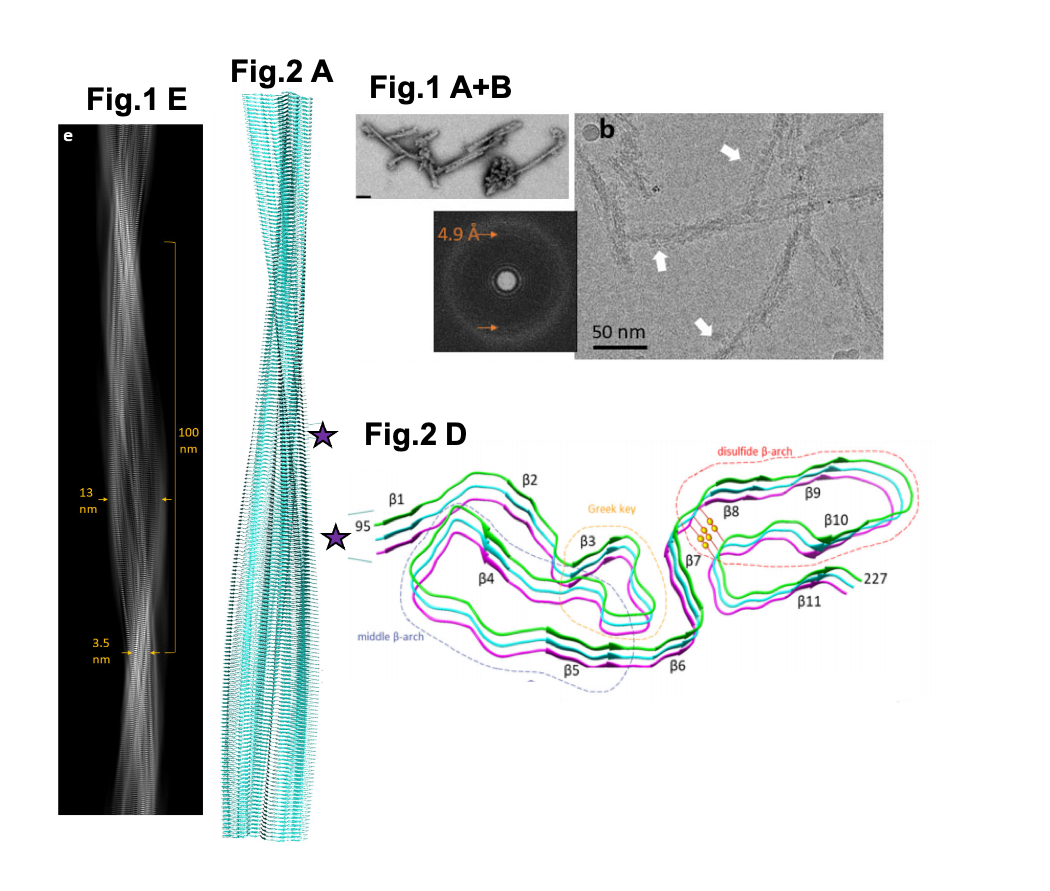
A selection of figures from Kraus et al. Fig. 1A. Negative stained TEM images of 263K prion fibrils (scale bar = 50 nm). Fig 1B. 2D cryo-EM images of 263K prion fibrils. Orange arrows = regular 4.9 Å spacing, white arrows = globules. Fig. 1E. A project of the fibril density map. Fig. 2A. An extended model of the fibril shown as a ribbon diagram. Fig. 2D. The stacking of beta-sheets within a region of the fibril. Different motifs are labelled and depicted by dashed lines. Yellow spheres = disulfide bond. Purple stars = position of the enlarged region on the fibril. Figures reproduced under a CC0 license.
- Using Cryo-EM, the authors produce a high-resolution structure (3.1 Å) of a mammalian infective prion. They solve the structure of an infectious prion isolated from hamster brains infected with the prion strain 263K, which was derived initially from sheep with the lethal neurodegenerative condition called Scrapie.
- The structure of 263K prion fibrils reveals their arrangement as left-hand twisted (>99%) fibrils between 13-20 nm in width at the widest point of the fibre cross-section and ~ 3.5 nm at the narrowest, in keeping with various prior studies on prion structure. From the start of one twist to another was ~ 100 nm. In ~ 45% of fibrils, globules were identified on one side of the fibril, sometimes spanning the entire length. The authors are unsure as to whether these structures are artifices occurring during sample preparation.
- The structure of the infectious prion 263K appears to be very different to that of synthetic recombinant human PrP amyloid fibrils, which have much smaller ordered cores and are therefore likely to be non-infectious.
- The authors resolved different motifs within the originally disordered N-terminal region, which were unable to be resolved in non-infectious PrPC fibrils from previous studies. These motifs included a beta-arch, and a Greek key motif (which is a serpentine fold containing beta sheets and loops). The C-terminal region had another beta-arch which, in this case, was disulfide-linked and heavily glycosylated. The authors suggest the combination of these features may contribute to the infectivity of prion 263K as one or more were absent in the non-prion PrP fibrils.
- They show that the location of the GPI anchors (at one side of the fibril) might help them to bind to membranes. This means the fibrils could be wrapped up in host membranes, which might contribute to pathognomonic features of prion pathology. The exposed (non-wrapped up) ends could be the site associated with refolding of the normal protein into the prion isoform.
To date, two major structural models have been proposed for infectious prion organisation, 1) arranged as a 4-rung beta solenoid like structure or 2) a Parallel in register intermolecular beta sheet (PIRIBS) where monomers are aligned and stacked.
- The authors showed that the 263K prion has a PIRIBS-based structure; further studies will be needed to determine if other prion strains have PIRIBS architectures as well, or in some cases, beta solenoids.
What I liked about this preprint:
I have always been fascinated by prion pathologies. It is interesting to think that misfolding of a protein can cause such devastating diseases. Given this, understanding what prions look like once they become pathogenic and how they become pathogenic by altering their shape is very important to understand how they arise and replicate. Cryo-EM is a great technique for this purpose. The author’s data shows they have been able to isolate a prion structure, build a high-resolution model, and reveal some peculiarities about the organisation of their prion contributing to our knowledge on prion structure.
Questions for the authors:
Q1: Prion fibrils can be very hard to isolate due to resistance of these proteins to proteases and detergents. What challenges did you face during the extraction and isolation of the 263K prion?
Q2: PrPSC is a highly glycosylated structure. It has been argued that such glycosylation could only be accommodated with beta-solenoid structures, but having now solved the structure of 263K at high-resolution, you show that 263K has a PIRIBS-based structure. Other mammalian prions, for example from mice, have been predicted (though not directly shown) to have a beta-solenoid-based architecture. Why do you think there is a difference between your prion structure and the beta-solenoid model hypothesised for murine prions? Do you think prion structure varies in this manner due to the host species? Or could it be previous studies have performed cryo-EM on anchorless molecules i.e does having the anchor drastically alter the structural configuration?
Q3: What is the role of the Greek key motif in this prion (is this known?) and how might this play a role in altering the conformation of PrPC to PrPSC?
References:
- Gitler, A.D., Dhillon, P., and Shorter, J. Neurodegenerative disease: models, mechanisms, and a new hope. Dis Model Mech. (2017).
- Collinge, J., and Clarke, A. A general model of prion strains and their pathogenicity. Science. (2007).
- Lodhi, S.K., Mirza, M., and Khawaja, A.M. Prions Disease: A review of novel transmission methods and efforts at discovering preventative and therapeutic modalities. Infectious Dis. In Clin Pract. (2018).
- Kulenkampff, K., Perez, A.M.W., Sormanni, P., Habchi, J., and Vendruscolo, M. Quantifying misfolded protein oligomers as drug targets and biomarkers in Alzheimer and Parkinson diseases. Rev. Chem. (2021).
- Mastriani, J.A. Chapter 21- Prion Diseases. Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease (Firth Edition). (2015).
- Murata, K., and Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. (2018).
doi: https://doi.org/10.1242/prelights.27565
Read preprint (No Ratings Yet)
(No Ratings Yet)Have your say
Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the biochemistry category:
Site-Specific Inhibition of Translation Initiation via 2’-O-methylation
Leonie Brüne
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the biochemistry category:
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
Peer Review in Biomedical Sciences
Communication of scientific knowledge has changed dramatically in recent decades and the public perception of scientific discoveries depends on the peer review process of articles published in scientific journals. Preprints are key vehicles for the dissemination of scientific discoveries, but they are still not properly recognized by the scientific community since peer review is very limited. On the other hand, peer review is very heterogeneous and a fundamental aspect to improve it is to train young scientists on how to think critically and how to evaluate scientific knowledge in a professional way. Thus, this course aims to: i) train students on how to perform peer review of scientific manuscripts in a professional manner; ii) develop students' critical thinking; iii) contribute to the appreciation of preprints as important vehicles for the dissemination of scientific knowledge without restrictions; iv) contribute to the development of students' curricula, as their opinions will be published and indexed on the preLights platform. The evaluations will be based on qualitative analyses of the oral presentations of preprints in the field of biomedical sciences deposited in the bioRxiv server, of the critical reports written by the students, as well as of the participation of the students during the preprints discussions.
| List by | Marcus Oliveira et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Cellular metabolism
A curated list of preprints related to cellular metabolism at Biorxiv by Pablo Ranea Robles from the Prelights community. Special interest on lipid metabolism, peroxisomes and mitochondria.
| List by | Pablo Ranea Robles |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
5 years
Jesús R. Requena
The beta solenoid model is based mostly on structural data and restraints obtained from mouse prions but it was proposed with the assumption that it is universal for all PrPSc prions. And the same can be said about the PIRIBS model. Both models competed in a “sine missione” fight for some time. The fact that the solenoid model could accomodate stacking of the glycans whereas apparently the PIRIBS could not, gave a powerful edge to the solenoid because it established a definitive physical restraint. Therefore, the nearly simultaneous publication, about a year ago, of studies by Byron Caughey’s and my group together with Biasini’s of modelling studies showing that PIRIBS can accommodate the glycans, was a hard blow to that advantage. And of course the cryo-EM study by Kraus et al puts things really difficult for the solenoid model, as now all the burden of evidence is on it. So, is it possible that some PrPSc strains are PIRIBS and some solenoids? Yes; but it is very unlikely because a prionic conformation is already a “one in a million” event. A protein able to fold into two of such conformations? I woud say it would be a one in a billion event…A careful revision of the structure of GPI-anchorless PrPSc using the higher resolution cryoEM techniques will be key. For starters, there is a feature in our reconstruction that has always bothered me a lot: the two intertwinned protofilaments are very thin and have a very large gap of “vacuum” between them. That does not make physical sense. So perhaps the “protofilaments” are actually bulges in a flat ribbon. Anyways, we’ll see…In the meantime, using PITHIRDS, we have just found that infectious recombinant PrPSc features a PIRIBS architecture:
https://doi.org/10.1101/2021.07.20.453078
Jesus Requena