








Target-specific precision of CRISPR-mediated genome editing
Posted on: 27 September 2018
Preprint posted on 9 August 2018
Article now published in Molecular Cell at http://dx.doi.org/10.1016/j.molcel.2018.11.031
The predictability of genome editing outcome varies across target sites and primarily depends on the nucleotide in the -4 position from the PAM site. Careful selection of target site is therefore key to inducing a specific desired modification.
Selected by Rob HyndsCategories: bioinformatics, molecular biology
Background
Cas9 is an RNA-guided DNA endonuclease that operates in the CRISPR (clustered regularly interspaced short palindromic repeats) bacterial adaptive immune mechanism. In these bacteria, short lengths of DNA from plasmids or bacteriophages are transcribed to CRISPR RNAs (crRNAs) which then provide the specificity for the Cas9 endonuclease to destroy the invading pathogen. Cas9 recognition of foreign DNA relies on crRNA, which contains a 20-nucleotide recognition region known as a protospacer, and a tracrRNA which hybridises with the crRNA. The role of the crRNA-tracrRNA complex can be performed by a single guide RNA (sgRNA) and this strategy is now widely used for genome editing in mammalian cells. The Cas9-sgRNA complex binds to homologous genomic DNA where aprotospacer adjacent motif (PAM) sequence (e.g. NGG for Cas9 from S. pyogenes) is present downstream of the target sequence. Cas9 induces a double-stranded break that is then repaired by endogenous DNA repair processes and can be further manipulated by inclusion of repair templates.
When no template for repair is provided, double-stranded breaks induced by Cas9 are repaired using error-prone repair pathways. These pathways introduce frameshift insertions or deletions (indwells) that disrupt the open reading frame and generate non-functional proteins, phenocopying gene knockout. A previous paper showed that the outcome of these repair events is not random and disruption of specific target sites can have a preferred repair outcome.
A current focus of research is finding ways to improve the design of sgRNAs to successfully target the locus of interest while minimizing off-target events elsewhere in the genome. In silico prediction tools now allow researchers apply our existing knowledge of the sequence patterns that correlate with high efficiency sgRNA activity but these remain imperfect.
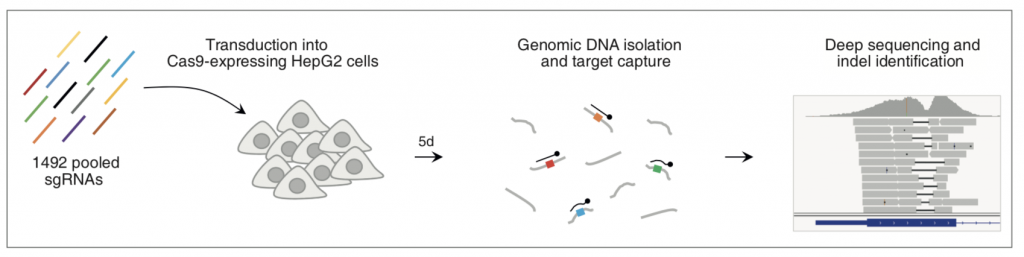
Figure 1A: Experimental Design
Key findings
In their preprint, Chakrabarti and colleagues examine the pattern of indels generated during CRISPR-Cas9-mediated gene editing in the absence of a repair template. The authors assessed repair of 1492 target sites in 450 genes in HepG2 cells using a pooled lentiviral library of sgRNAs predicted to have high activity and confirm that the outcome of editing is non-random in biological replicates. Single nucleotide (nt) indels occurred most frequently but there was a long tail in the length of indels and the preferred indel length for some target sites was as long as 56 nt. Almost 90% of indels produced frameshift but some target sites showed in-frame indel preference, suggesting that they should be avoided for gene KO studies. This, along with the observation that multiple sgRNAs seemed to have lower activity than predicted suggests that prediction algorithms can be further refined through better understanding of the activity of Cas9 in human cells. The pattern of indels at different sites varied with some having one strong preference and others having little preference between dozens of possibilities; the finding that only one-fifth of target sites (‘precise targets’) have a greater than 50% probability of inducing one specific indel is significant as predicting the specific outcome of genome editing for the remaining target sites is not easy.
Further characterisation of precise targets revealed that editing at these was more efficient. Precise targets were more likely to be insertions and more likely to be a single nucleotide in length while imprecise targets favoured deletions. Microhomology around the indel appeared to be a feature of deletions, consistent with repair by the MMEJ pathway. Some single nucleotide insertions also showed a preference for a common base suggesting that the nucleotide choice is not random. The preferred base was homologous to nucleotide -4 from the PAM sequence, which is usually one nucleotide upstream of the cleavage site. Moreover, precise targets showed base preference at the -4 position: when the target has an “A” or a “T” in the -4 position, repair is likely to result in a highly recurrent insertion but when it is a “G”, deletions are more likely and repair is less predictable.
These data clarify the role of DNA sequence in repair precision after Cas9 cleavage but the failure of algorithms based solely on sequence to predict indel profiles accurately suggests that other factors might influence the indel profiles of target sites. In this regard, the preprint finds a role for chromatin structure. Addition of a HDAC inhibitor or an EZH2 inhibitor altered the indel profiles observed for the same target sites by increasing and decreasing indel formation, respectively. The extent of these changes was similar to when DNA repair pathways are pharmacologically manipulated supporting the importance of chromatin structure. Some changes in the relative frequency of specific indels at targets sites were observed but were broadly similar to the untreated conditions. That said, the authors demonstrate that for some target sites, altering the chromatin state can change the most frequent indel and favour some indels over others. Future work should address the hypothesis that there is more complex interplay between chromatin state and DNA repair pathway choice than is currently appreciated.
Conclusions
- DNA sequence features affect the indel profiles generated after CRISPR-Cas9 gene editing.
- Chromatin structure also affects indel formation and inducing histone acetylation improves the efficiency of editing.
- Establishing and targeting precise sites will maximize the likelihood of desirable outcomes in experimental and clinical applications of gene editing.
Further Reading
Taheri-Ghahfarokhi, A., Taylor, B. J. M., Nitsch, R., Lundin, A., Cavallo, A. L., Madeyski-Bengtson, K., Karlsson, F., Clausen, M., Hicks, R., Mayr, L. M. et al.(2018). Decoding non-random mutational signatures at Cas9 targeted sites. Nucleic Acids Res 46, 8417-8434.
Allen, F.R., Crepaldi, L.R., Alsinet-Armengol, C. ,Strong, A., Kleshchevnikov, V., Pietro De Angeli, P., Palenikova, P., Kosicki, M., Bassett, A.R., Harding, H. et al.(2018). Mutations generated by repair of Cas9-induced double strand breaks are predictable from surrounding sequence. bioRxiv.
Questions for Authors
Q1. The finding that the chromatin state of a particular target influences targeting efficiency is interesting as it is not obvious how you could incorporate this into existing prediction algorithms as the optimal sgRNA to knockout a gene in two different cell types is likely to be different. Do you think the indel patterns observed would be consistent in other cell lines, for example?
Q2. What are the implications for the progression of CRISPR-based technologies to the clinic if editing of the same target can vary between cell types or even between the same cell type in two patients?
Q3. Your preprint caused a stir on Twitter as it acknowledges Her Majesty Queen Elizabeth II for starting your sequencing run. Could you explain a bit more?
doi: https://doi.org/10.1242/prelights.4951
Read preprint (2 votes)
(2 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the molecular biology category:
Aging increases ovarian cancer growth, metastasis, and immunosuppression that can be alleviated by inhibiting hedgehog signaling
Zoha Sadaqat
A pre-rRNA positive feedback loop drives malignant ribosome biogenesis
Vaishali Grewal
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |