








A pharmacokinetic-pharmacodynamic assessment of the hepatic and bone-marrow toxicities of the new trypanoside fexinidazole
Posted on: 18 December 2018 , updated on: 29 September 2019
Preprint posted on 4 December 2018
g-HAT tricks: linking fexinidazole’s liver and bone marrow toxicities to patients’ exposure to its bioactive metabolite
Selected by Zhang-He GohCategories: pharmacology and toxicology
Background of preprint
Fexinidazole, a small molecule containing the nitroimidazole scaffold, has been identified as a potential new treatment for human African trypanosomiasis caused by Trypanosoma brucei gambiense (g-HAT), as well as for Chagas disease. Given the fatality of these diseases and the lack of safe and effective treatments, alternative therapies are urgently needed. First reported by Torreele et al. in 2010 [1], fexinidazole is a promising new treatment given its efficacy against both the blood and central nervous system stages of g-HAT. Unfortunately, progress on taking fexinidazole into the clinic stalled briefly when serological markers measured were suggestive of liver and bone marrow toxicities.
Therefore, Watson et al. had two aims in their study:
- To characterise the relationships between the exposure to fexinidazole and its metabolites, and the potential adverse effects.
- To predict safety of the ten-day fexinidazole regimen developed for g-HAT treatment.
To fulfil the first aim, Watson et al. modelled the formation of the fexinidazole sulfone metabolite M2 as a “first-order absorption” process because fexinidazole is metabolised in vivo to two biologically active metabolites, M1 and M2 (Fig. 1); the latter is a good marker for bioactive exposure for the ten-day treatment of g-HAT using fexinidazole. The authors then made various adjustments to their model in the prediction of this ten-day fexinidazole treatment.
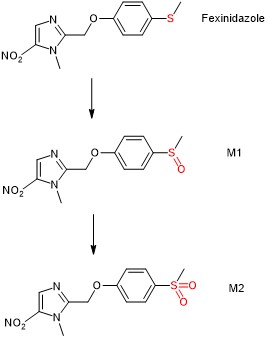
Figure 1. Metabolism of fexinidazole to biologically active M1 and M2.
Key findings of preprint
To estimate the exposure to fexinidazole, Watson et al. used a one-compartment disposition model with two transit compartments for M2 formation. The authors also included inter-individual random variability in pharmacokinetic (PK) parameters to improve model fit. Having established the model, Watson et al. then linked liver and bone marrow toxicities to M2 concentrations observed or predicted in the model.
(A) Liver toxicity
Watson et al. analysed data acquired from a previous dose ranging study of fexinidazole used in the treatment of symptomatic Chagas disease patients. The authors used fexinidazole exposure as the predictive variable, and haematological timeseries and liver timeseries as outcome variables. *
From the data summarised in Figure 2 of their paper, the authors made two observations. First, the time-series data exhibited considerable heteroscedasticity. In the context of this paper, this means that the variability of liver transaminase elevation was unequal across the duration of fexinidazole exposure. Second, the authors noted a visible distinction of liver transaminase elevation between patients with high and low M2 exposures, which could be ascribed to total drug exposure.
The authors found that their model revealed a clear relationship between fexinidazole exposure and the outcome of hepatotoxicity, as measured using the both the absolute and relative elevations of liver transaminases. This trend predicted that approximately 50% of individuals with M2 exposures as distributed in the three studies for the g-HAT regimen would experience ALT and AST elevations more than 2.8 times and more than 2 times the baseline respectively; however, no such cases were observed.
(B) Bone marrow toxicity
Using the models constructed on indicators of bone marrow toxicity, Watson et al. found clear exposure-response relationships between M2 exposure and bone marrow toxicity. Specifically, the authors used neutrophil and platelet counts as indicators. In contrast, both the absolute and relative models measuring lymphocytes and haemoglobin levels did not appear to exhibit an exposure-response effect. This led Watson et al. to conclude that the decreases in neutrophil and platelet counts were predictable.
What I like about this preprint
I selected this preprint for its significance in characterising the relationship between fexinidazole’s PK and pharmacodynamic (PD) properties—this preprint focuses on overcoming the current difficulties that must be resolved so that fexinidazole can be used on a wider scale for g-HAT and Chagas disease. Moreover, in highlighting this preprint, I hoped to shed some light on the difficulties associated with predicting the toxicity of a potential drug.
Three such challenges were discussed in this preprint:
- All models run the risk of oversimplification, overfitting, and confounding. This difficulty arises from the maxim that statistical analyses are only as robust as the data that they are built on. In this study, Watson et al. pointed out that the patients suffering from g-HAT were symptomatic, and implied that they may have had higher baseline neutrophil and platelet counts on admission compared to their asymptomatic counterparts in the Chagas study.
- A certain amount of uncertainty may be attributed to the data used in the model. This second point may be associated with the practical limitations of study design. In their model, Watson et al. used M2 AUlogC as an proxy indicator for fexinidazole exposure; this meant that their analysis was limited to drawing associations between M2 exposure and the toxic outcomes experienced by the participants in the trials.
- The applicability of findings generated from experiments and modelling depends heavily on both their statistical and clinical significance. Researchers may sometimes generate statistically significant findings, but these findings must also have pertinent clinical implications in order to be adopted into medical practice. This led Watson et al. to postulate that “the fexinidazole regimen for g-HAT treatment results in mild but predictable delayed reversible decreases in neutrophil and platelet counts”.
Therefore, seen from this perspective, this preprint serves as a useful synthesis in bringing together data from previous clinical studies. In general, clinical studies require tremendous amounts of effort and resources to conduct, which means that they differ greatly in study design depending on the investigators’ resources and constraints. In conducting this research, Watson et al. face the mammoth task of reconciling apparent differences in these clinical studies and clinical trials, assess the prevailing literature, and recommend directions for future research.
Future directions
Based on their findings in this preprint, Watson et al. concluded that the margin of safety for dose-related toxicity is enough to justify the use of fexinidazole in the treatment of g-HAT. Future directions will likely focus on establishing a firmer picture of fexinidazole’s safety profile, which may extend to elucidating its dose-dependent mechanisms of toxicity using in vitro cell-based and in vivo animal-based experiments. At the same time, the potential that fexinidazole exhibits idiosyncratic toxicity must also be considered. Such a possibility may be investigated by assessing clinical symptoms or markers characteristic of immune-mediated reactions to fexinidazole, such as antibody production.
On the other side of the fence, fexinidazole’s PK profile will be further studied. Better assays may be developed that allow researchers to sensitively and specifically measure for concentrations of fexinidazole, M1, and M2. The ability to measure the concentrations of these three moieties in other body fluids such as urine or bile will also serve to propel the understanding of fexnidazole’s PK forward.
Questions for authors
- In Table 2, the OVL of the absolute model for lymphocytes and haemoglobin were 69 and 75 respectively; and the OVL of the relative model for lymphocytes and haemoglobin were 11 and 29 respectively. Similarly, the OVL of the absolute model for platelet EC50 and ALT were 3 and 2 respectively. Previously, in the writeup for Table 2, the OVL appeared to be limited to a range of values between 0 and 1. Therefore, could you elaborate on how the abovementioned OVL values should be interpreted in this context?
References
[1] Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazué G, Bray MA, Pécoul B, Fexinidazole – A New Oral Nitroimidazole Drug Candidate Entering Clinical Development for the Treatment of Sleeping Sickness, PLOS Neglected Tropical Diseases 4(12) (2010) e923.
Corrigendum
* This part of the preLight originally read:
Three variables were discussed: (i) liver transaminase levels, a marker of hepatotoxicity; (ii) time series; and (iii) fexinidazole exposure, as estimated using M2 exposures.
The authors have pointed out that this is not exactly correct. Their comments can be read in the author response.
The preLight author is immensely grateful to the preprint authors for their editing suggestions and feedback.
doi: https://doi.org/10.1242/prelights.6556
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the pharmacology and toxicology category:
The Endocannabinoid System’s Contribution to Placebo Analgesia
Thomas Nicodemo Arrieta et al.
Small Molecule Agonists of TREM2 Reprogram Microglia and Protect Synapses in Human Alzheimer’s Models
Dina Kabbara
Snake venom metalloproteinases are predominantly responsible for the cytotoxic effects of certain African viper venoms
Daniel Osorno Valencia
preLists in the pharmacology and toxicology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
COVID-19 / SARS-CoV-2 preprints
List of important preprints dealing with the ongoing coronavirus outbreak. See http://covidpreprints.com for additional resources and timeline, and https://connect.biorxiv.org/relate/content/181 for full list of bioRxiv and medRxiv preprints on this topic
| List by | Dey Lab, Zhang-He Goh |
1
Drug use in special populations
Any drugs that are being used in special populations: Patients with liver and kidney failure, in paediatrics, in geriatrics, and in pregnant or lactating patients. Includes the discovery of factors that could potentially affect drug use in these special populations.
| List by | Zhang-He Goh |
Toxicology of toxicants, existing therapeutics, and investigational drugs
Preprints that describe the toxicology of environmental pollutants and existing and upcoming drugs. Includes both toxicokinetics and toxicodynamics, as well as technological improvements that will help in the characterisation of this field.
| List by | Zhang-He Goh |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Advances in Drug Delivery
Advances in formulation technology or targeted delivery methods that describe or develop the distribution of small molecules or large macromolecules to specific parts of the body.
| List by | Zhang-He Goh |