








Mixed Alkyl/Aryl Phosphonates Identify Metabolic Serine Hydrolases as Antimalarial Targets
Posted on: 2 February 2024 , updated on: 8 October 2024
Preprint posted on 11 January 2024
Article now published in Cell Chemical Biology at http://dx.doi.org/10.1016/j.chembiol.2024.07.006
Lessons from nature for antimalarial agent discovery: @ChemBio_John and colleagues (from @mbogyo and @fidocklab) develop serine hydrolase inhibitors targeting plasmodium parasites. Preprint highlighted by @goh_zhanghe.
Selected by Zhang-He GohCategories: microbiology, pharmacology and toxicology
Updated 8 October 2024 with a postLight by Zhang-He Goh
This highlighted preprint has now been published in Cell Chemical Biology.
In this work, Bennett and co-workers from the Bogyo group developed a series of mixed alkyl/aryl phosphonates that exhibit antimalarial properties inspired by SalA. They investigated the structure-activity relationship of these molecules, leading them to identify their most potent antimalarial compound. Interestingly, its mechanism of action has not been fully investigated, but further experiments by the authors show that (a) it must be distinct from SalA and the pan-lipase inhibitor Orlistat, and (b) it inhibits multiple targets including a serine hydrolase (abH112).
I did not notice any major changes between the preprint and the published version. The authors have added a Limitations section in the published version, in which they describe the need to better understand the impact of serine hydrolases (the targets of the authors’ antimalarial compounds) in parasite survival. This would in turn allow to researchers to better understand the mechanism of action of these inhibitors.
Congratulations again to the authors.
Background of the preprint
Malaria is a highly transmissible disease afflicting hundreds of millions of people worldwide. It is caused by various Plasmodium parasites. One of these parasites, Plasmodium falciparum, is implicated in about half of all malaria cases. Worse still, it causes one of the most dangerous forms of malaria. Currently, resistance to antimalarial treatments targeting P. falciparum is rapidly growing. Given malaria’s widespread disease burden and its threat to global public health, there is an urgent need to develop antimalarial treatments with alternative mechanisms of action.
The search for effective antimalarial agents targeting P. falciparum has been greatly inspired by natural product discovery. In a series of highly collaborative efforts, the Bogyo and Fidock groups have ascribed the antimalarial activity of one such product, Salinipostin A (SalA), to its ability to inhibit various serine hydrolases.1 Serine hydrolases play key roles in lipid metabolism and in maintaining P. falciparum’s ability to remodel its plasma membrane throughout its life cycle in the host’s red blood cells.
The upshot of SalA having multiple targets is that resistance to SalA is unlikely to develop. However, the synthesis and isolation of SalA is too challenging for SalA to be a viable therapeutic option in the treatment of malaria. Therefore, the Bogyo and Fidock groups aimed to develop a class of antimalarial agents that would be easier to make. To this end, Bennett and co-workers developed a series of mixed alkyl/aryl phosphonates, tested their structure-activity relationship, and characterised their activity on both purified enzymes and on P. falciparum parasites (Figure 1).
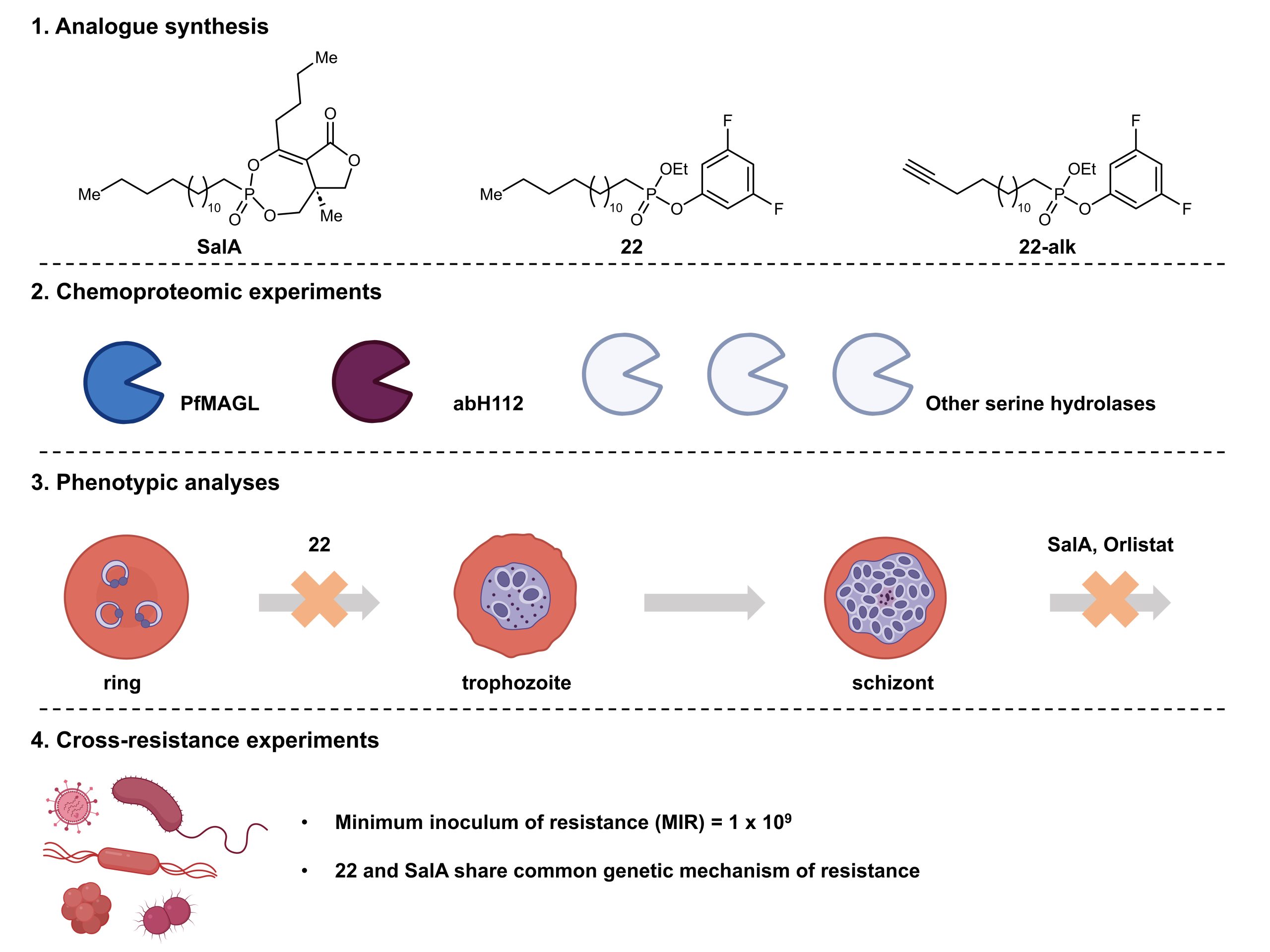
Figure 1. Summary of this work by Bennett and co-workers.
Key findings of this preprint
(A) Synthesis of phosphonates
Given that the key difficulty in the synthesis of SalA and its analogues lies in the synthesis of the bicyclic enolphosphate group, Bennett and co-workers first developed a synthetic strategy so that they could rapidly access a series of mixed alkyl/aryl phosphonates. The preprint authors then ran a series of biochemical assays to test these mixed alkyl/aryl phosphonates against a serine hydrolase target of SalA named PfMAGL. These experiments proved that the alkyl/aryl phosphonates were potent inhibitors of PfMAGL and showed that electron-withdrawing substituents on the phosphonate phenols were beneficial for inhibitory activity while electron-donating and sterically bulky substituents were not. Unfortunately, the authors also found that these compounds did not exhibit potent antiparasitic properties over 72 hours.
In medicinal chemistry, the structure of a compound is closely linked to its pharmacological activity. Therefore, the authors next synthesised a second series of analogues which more closely resemble SalA with the goal of better preserving its antimalarial activity. This strategy was successful, as further experiments led Bennett and co-workers to identify an analogue, labelled compound 22 in the preprint, as a potent inhibitor of parasite growth. Compound 22 exhibited excellent inhibitory activity in both biochemical and parasitic assays; it also exhibited minimal mammalian cell toxicity, so the authors focussed on compound 22 in subsequent experiments.
Bennett and co-workers performed chemoproteomic experiments to confirm the targets of the analogues that they had synthesised. To do this, the authors made activity-based probe versions of these analogues, including that of compound 22 (labelled compound 22-alk in the preprint). The authors found that compound 22 inhibited multiple serine hydrolases including PfMAGL, a known target of SalA. In fact, the antimalarial activity of compound 22 did not arise from the inhibition of PfMAGL alone; other serine hydrolases including AbH112 were implicated as well.
(B) Phenotypic analyses
Next, Bennett and co-workers performed phenotypic analyses, which showed that compound 22 blocks P. falciparum’s life cycle at a different stage compared to both SalA and the pan-lipase inhibitor Orlistat. This suggested that compound 22 has a different mechanism of action compared to both SalA and Orlistat. Specifically, compound 22 blocks the parasites’ transition from the late ring to the early trophozoite stage. In contrast, both SalA and Orlistat block the parasites’ progression at the late schizont stage.
(C) Antimalarial resistance experiments
Finally, the authors performed cross-resistance experiments between compound 22 and SalA. Interestingly, despite their distinct mechanisms of action, antimalarial resistance against compound 22 implicated mutations similar to those which conferred antimalarial resistance against SalA. These findings led the authors to postulate that compound 22 and SalA may share a common genetic mechanism of resistance. In these experiments on resistance, compound 22 had a minimum inoculum of resistance of 1 x 109, leading the authors to conclude that it is less prone to induce resistance than other antimalarials.
Given that the authors previously identified AbH112 as a possible target, they also examined the role of AbH112 in parasite growth. Interestingly, they found that conditionally knocking down the expression of AbH112 did not significantly affect the growth or development of P. falciparum, nor did it affect its susceptibility to compound 22.
What I like about this preprint
Antimalarial treatment is a challenging but important research area—I have written about it twice since joining the preLights community in 2018, and would like to highlight it again with my first preLight of 2024. Key advances in chemical biology in recent years have given researchers multiple tools to A) rapidly synthesise complex molecules that were previously deemed difficult or impossible to produce and B) use them to quickly identify their mechanisms of action. This has allowed us to better identify new biological targets for the treatment of complex diseases.
Indeed, covalent inhibitors are making a comeback. Previously, covalent inhibitors were deemed difficult to work with because researchers were concerned about their lack of selectivity—and by extension, increased toxicity. However, researchers are now much better equipped to invent inhibitors specific for their target.2
In this preprint, Bennett and co-workers demonstrate how covalent inhibition can be useful, even when implicated in multiple mechanisms of action. In trying to identify the exact mechanism of action underpinning their SalA-inspired covalent inhibitors, the authors showed that these inhibitors bind to various proteins that have previously been implicated in antimalarial activity. It would be exciting if these findings could ultimately lead to the invention of agents with improved activity, reduced risks of resistance, and reduced toxicity.
Acknowledgements
Images created using Microsoft Powerpoint, ChemDraw, and BioRender.
Questions for authors
- Could you describe how you designed the SalA analogue library? Specifically, how did you select the phenol leaving groups (as opposed to other leaving groups)? Did you also consider other alkyl substituents, such as cyclic alkyl structures, on the SalA analogues?
- Given that SalA and its analogues act through a covalent mechanism, do you think the covalent inhibition is irreversible in all cases? Is it possible that the covalent inhibition is more reversible on some enzymes than others?
- In the same vein, how quickly does the covalent inhibition occur? Is the inhibition kinetics of these agents (in this case, the phosphonate group) rapid?
References
- E. Yoo, C. J. Schulze, B. H. Stokes, O. Onguka, T. Yeo, S. Mok, N. F. Gnädig, Y. Zhou, K. Kurita, I. T. Foe, S. M. Terrell, M. J. Boucher, P. Cieplak, K. Kumpornsin, M. C. S. Lee, R. G. Linington, J. Z. Long, A.-C. Uhlemann, E. Weerapana, D. A. Fidock and M. Bogyo, Cell Chemical Biology, 2020, 27, 143-157.e145.
- L. Boike, N. J. Henning and D. K. Nomura, Nature Reviews Drug Discovery, 2022, DOI: 10.1038/s41573-022-00542-z.
doi: https://doi.org/10.1242/prelights.36398
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
Also in the pharmacology and toxicology category:
The Endocannabinoid System’s Contribution to Placebo Analgesia
Thomas Nicodemo Arrieta et al.
Small Molecule Agonists of TREM2 Reprogram Microglia and Protect Synapses in Human Alzheimer’s Models
Dina Kabbara
Snake venom metalloproteinases are predominantly responsible for the cytotoxic effects of certain African viper venoms
Daniel Osorno Valencia
preLists in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the pharmacology and toxicology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
COVID-19 / SARS-CoV-2 preprints
List of important preprints dealing with the ongoing coronavirus outbreak. See http://covidpreprints.com for additional resources and timeline, and https://connect.biorxiv.org/relate/content/181 for full list of bioRxiv and medRxiv preprints on this topic
| List by | Dey Lab, Zhang-He Goh |
1
Drug use in special populations
Any drugs that are being used in special populations: Patients with liver and kidney failure, in paediatrics, in geriatrics, and in pregnant or lactating patients. Includes the discovery of factors that could potentially affect drug use in these special populations.
| List by | Zhang-He Goh |
Toxicology of toxicants, existing therapeutics, and investigational drugs
Preprints that describe the toxicology of environmental pollutants and existing and upcoming drugs. Includes both toxicokinetics and toxicodynamics, as well as technological improvements that will help in the characterisation of this field.
| List by | Zhang-He Goh |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Advances in Drug Delivery
Advances in formulation technology or targeted delivery methods that describe or develop the distribution of small molecules or large macromolecules to specific parts of the body.
| List by | Zhang-He Goh |