








ATM-dependent formation of a novel chromatin compartment regulates the Response to DNA Double Strand Breaks and the biogenesis of translocations
Posted on: 17 February 2022
Preprint posted on 8 November 2021
Categories: molecular biology
Background:
The physical arrangement of chromatin within the nuclear space plays an important role in mediating many DNA-associated processes. For example, gene expression regulation, DNA duplication and DNA repair (1). Broadly, chromosomes are segregated into two ‘compartments’ known as ‘A’ (a gene dense and transcriptionally active) and ‘B’ (repressed chromatin). These regions are sub-divided into topologically associated domains (TADs), reflecting the level of contact between DNA regions. DNA regions within a TAD interact more frequently with each other than with DNA regions in other TADs. Within TADs the chromatin can physically ‘loop’ providing an additional structural layer (1).
We understand less about how chromatin organisation in the nucleus contributes to the cellular response to DNA injuries, which can drive instability in the genome.
Double strand breaks (DSBs) arise when both strands of the DNA helix break. If un-repaired, they can result in rearrangements, translocation events or the loss of DNA sequences thereby compromising the genome. At the heart of DSB repair are the kinases (proteins which transfer information to other proteins via phosphorylation), Ataxia-telangiectasia Mutated (ATM), and DNA protein kinase (DNA-PK). Both kinases are essential for orchestrating repair and preparing the chromatin environment by phosphorylating a variant histone H2AX (in mammals) to form yH2AX and subsequently recruiting other repair factors (2). Decades of work have characterised in detail DSB repair, though we are yet to understand why DSBs ‘cluster’ within the nucleus, forming sites of DNA repair, as this organisation could potentially promote more instability. For example, during the process of repair, DNA ends could be incorrectly joined if they are close together (3).
Here, the authors investigate how the chromatin structure alters in the presence of DSBs. Their work now supports the presence of a new G1-phase and DSB specific chromatin compartment (the ‘D’ compartment). They suggest that this compartment, which is associated with DSB clusters, could activate, and concentrate some DNA damage associated factors thereby generating an optimal repair response/environment to tackle DSBs. However, they show that such compartmentalisation may also account for some genome instability events seen in cancerous cell lines.
Key Findings:
Here, the authors use a human cell line they developed called DIvA cells. DIvA (DSB Inducible via AsiSI) cells can be induced to produce ~ 100 DSBs using an endonuclease called AsiSI. Adding OHT (hydroxytamoxifen) induces the movement of the AsiSI endonuclease from cytoplasm to nucleus, where it then cuts the DNA (4). To assess changes in chromatin structure after a DSB, the authors performed chromatin conformation capture experiments including Hi-C and 4C-seq. Briefly, Hi-C preserves interactions between different regions of DNA via cross-links in an unbiased manner. Once the cross-linked DNA is digested, biotinylated nucleotides are incorporated at DNA ends. The fragments are then ligated together and after, the crosslinks are removed. This results in the formation of a hybrid DNA fragment containing DNA regions that are distant linearly in the genome but that were close physically in the nuclear space at the beginning of the experimental procedure. Using streptavidin (which binds biotin), these hybrid DNA fragments are isolated then analysed to measure the interaction frequency between these distant genomic loci (5). 4C-seq follows a similar concept but allows the detection of all potential sequences which are interacting with a specific DNA region of interest (6).
A) Damaged TADs are supported by ATM activity
Performing Hi-C in DIvA cells in the presence and absence of ATM kinase activity, revealed that if ATM activity was present, TADs containing DSBs were less in contact physically with other (non-DSB containing) TADs. In some cases, the DSB itself became isolated within the TAD. Whereas, inhibiting ATM activity in the presence of DSBs using an ATM-specific kinase inhibitor (KU-55933; 7), the authors found less interactions occurring within TADs and more limited spreading of yH2AX signal. They also noted that blocking ATM activity prevents DNA loop extrusion, which the authors previously demonstrated as a driver of DSB repair foci. Loop extrusion is a process by which chromatin ‘loops’ grows via the action of cohesin (8). In contrast, by inhibiting DNA-PK, which impair severely DSB repair, using a kinase inhibitor (NU-7441), they found increased intra-TAD interactions, accumulation of yH2AX and increased loop extrusion.
Thus, ATM activity appears to promote isolation of the DSB damaged region and can induce alterations to chromatin structure.
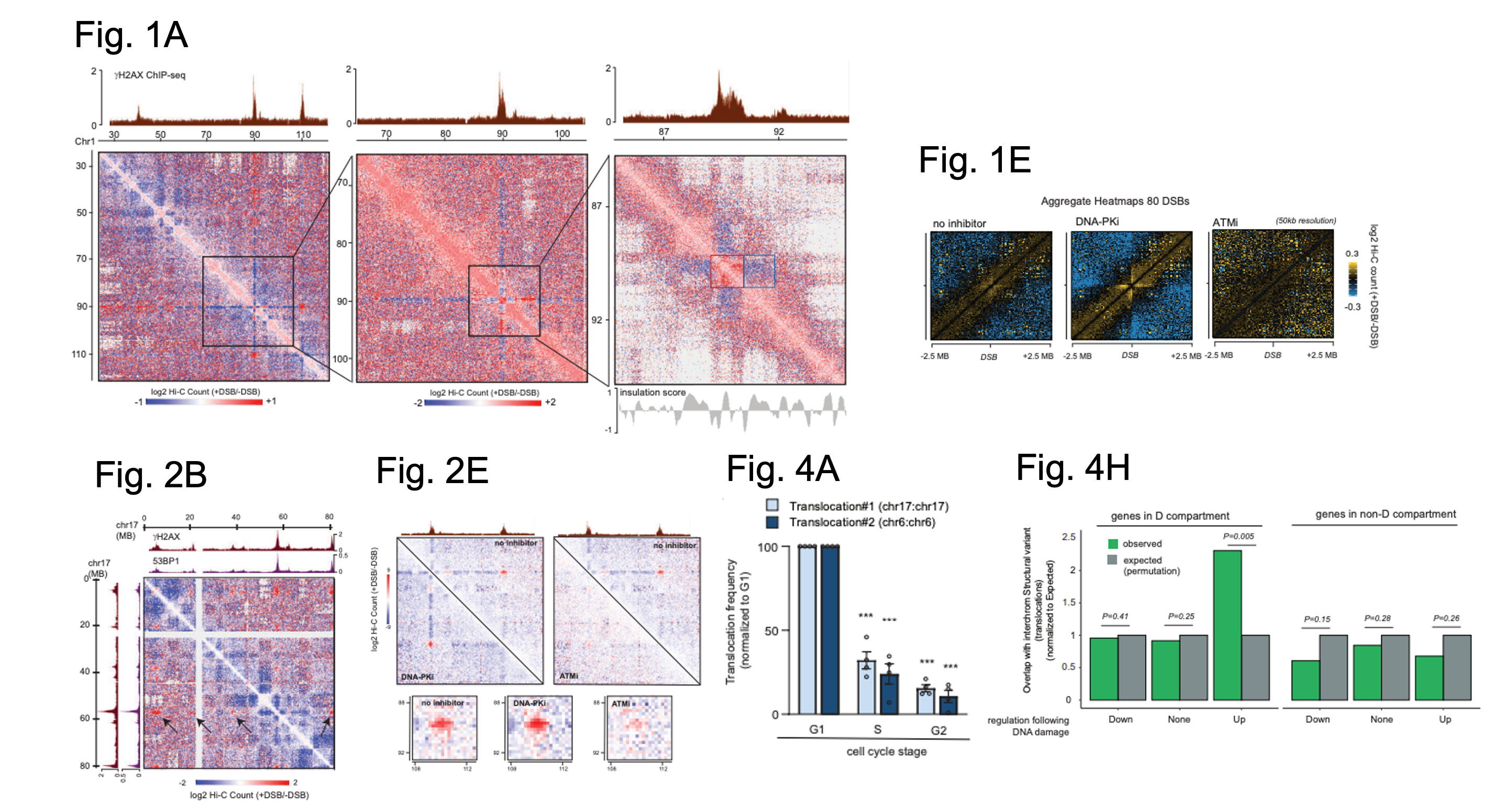
Figure shows a selection of figures from Arnould et al. Figure 1A shows Hi-C data from DIvA cells in the presence or absence of DSBs for part of chromosome 1 at three resolutions: 250 kb (left), 100 kb (middle) and 25 kb (right). The top tracks show the locations of yH2A signal indicating where a DSB has occurred. Red square (a damaged TAD with enhanced cis interactions), Blue square (decreased interaction between damaged TAD and nearby TAD. Figure 1E shows Hi-C contact matrices in the presence or absence of DSBs in control cells (left) and cells in which either DNA-PK (middle) or ATM (right) have been inhibited. Figure 2B shows Hi-C data in the presence or absence of DSBs on part of chromosome 17. The tracks on top and left show ChIP-seq defined locations of yH2AX and 53BP1. Black arrow = clustering of one DSB with several others on chromosome 17. Figure 2E shows Hi-C data as above (for a region on chromosome 1) but with the additional inhibition of either DNA-PK or ATM. Top tracks show yH2AX signal as defined by ChIP-seq. Boxes below show Hi-C between two yH2AX domains for each condition. Figure 4A shows qPCR data to assess translocation frequency for two different translocations after DSBs were induced in synchronised cells (as detailed on the graph). Figure 4H shows sites in which there are overlaps between sites of translocation events in cancer cells and genes associated with the D compartment. Expected overlap (grey, computationally defined), observed overlap (green). Figures adapted from Arnould et al., and made available under a CC-BY 4.0 International license.
B) DSB clustering generally happens during G1-phase of the cell cycle
The author’s data suggests that TADs containing a DSB can form close contacts (i.e cluster) with other DSB containing TADs. To find out what chromatin features were associated with these multiple TAD sites, they found yH2AX signal, other DSB associated chromatin marks (i.e loss of H1 histone, 53BP1 accumulation) and RNA polymerase II (RNAPII), all of which were determined via chromatin immunoprecipitation. Thus, DSB-associated/DNA repair factors are found at DSB clusters and these clustered DSBs came from sites of active transcription. By inhibiting ATM as before, the authors show that the clustering behaviour requires active ATM (but not DNA-PK whose inhibition enhanced the process). Lastly, the authors found that in synchronised cells (using thymidine which arrests cells at the G1/S transition), this clustering pattern appeared more prominently in G1 stage cells.
C) The ‘D’ compartment – a DSB clustering site
As mentioned, chromatin can be compartmentalised into ‘A’ (active) and ‘B’ (repressed) compartments. Though DSBs did not appear to affect this organisation nor did inhibiting ATM or DNA-PK, they saw that clustering was more common when a DSB happened within the ‘A’ compartment. Upon further investigation of their data, the authors uncovered an additional ‘A’ associated sub-compartment, referred to as the ‘D’ (DSB-induced) compartment. They found this sub-compartment to be rich in yH2AX signal however some TADs associated with this sub-compartment did not appear to be yH2AX containing but were instead enriched for other chromatin marks associated with active transcription.
D) The D compartment – a hub of DSB repair?
Some undamaged TADs are associated with the ‘D’ compartment but why is unclear. To understand this behaviour, the authors examine for both DNA motifs and performed RNA-seq analysis in the presence and absence of DSB induction to look for gene expression changes associated with the ‘D’ compartment. They found that some tumour suppressor genes and DNA damage response factors were up-regulated, and associated with the ‘D’ compartment. When they disrupted the cell’s ability to cluster DSBs (via RNAi depletion of a factor called SUN2), they found reduced up-regulation of these genes via qPCR. Specifically, they examined for change in PLK3 (polo-like kinase3), RNF19B (ring finger protein 19B) and GADD45A (growth arrest and DNA damage inducible alpha), all of which are associated with DNA repair/tumour suppression responses. Thus, recruitment of these genes to the ‘D’ compartment could aid activation.
E) DSB clustering can result in translocations
A key negative consequence of DSB clustering is the potential for chromosome translocations (where chromosome/break ends are joined incorrectly). To ask about the frequency of translocations after DSBs, the authors used qPCR and Amplicon-seq assays (amplification and sequencing of DSB ends). This allowed to test for intra-chromosomal (i.e happening on the same chromosome) and inter-chromosomal translocations (i.e between different chromosomes). In keeping with ‘D’ compartment predominance in G1, the authors found that more translocations were happening during this cell cycle stage. If the authors enhance ‘D’ compartment formation (by blocking DNA-PK) they find increased translocations, whereas disrupting DSB clustering reduced translocations, suggesting that DSB clustering can lead to both intra and inter-chromosomal translocations. Interestingly, by limiting loop extrusion actors, they found less intra-chromosomal translocations, suggesting loop extrusion also plays a role in these events.
As genome instability, including translocations, are common hallmarks of cancer cells, the authors also investigated if some translocations occurring in cancer cells could be associated with the clustering of DSBs and DNA damage response genes in the ‘D’ compartment. Taking a computational approach, they demonstrated a correlation between genes associated with the ‘D’ compartment following a DSB with points where translocations were observed in cancerous genomes indicating that some translocation events could be perpetuated by events occurring within the ‘D’ compartment.
What I liked about this preprint:
I really like this study as I think it demonstrates well the power of mining next generation sequence data sets to retrieve novel and exciting biology. Here the authors combine some of their prior observations and across the field to address a key gap in our understanding of DSB behaviour. Though more work is needed to really understand the fine aspects of the clustering process, the results here now open a new avenue of investigation.
References:
- Szabo, F. Bantignies and G. Cavalli. Principles of genome folding into topologically associating domains. Science Advances. 2019.
- Scully, A. Panday, R. Elango and N.A. Willis. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Molecular Cell Biology. 2019.
- Marnef and G. Legube. Organizing DNA repair in the nucleus: DSBs hit the road. Current Opinion in Cell Biology. 2017.
- https://www.legubelab.com/research
- Kong and Y. Zhang. Deciphering Hi-C: from 3D genome to function. Cell Biology and Toxicology. 2019.
- H.L. Krijgerm G. Geeven, V. Bianchi, C.P.E. Hilvering and W. de Laat. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods. 2020.
- Hickson, Y. Zhao, C.J. Richardson, S.J. Green, N.M. Martin, A.I. Orr et al. Identification and characterisation of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Research. 2004.
- Arnould, V.Rocher, A-L. Finoux, T. Clouaire, K. Li, F. Zhou, P. Caron, P.lE. Mangeot, E.P. Ricci, R. Mourad, J.E. Haber, D. Noordermeer and G. Legube. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature. 2021.
- Aymard, M.Aguirrebengoa, E.Guillou, B.M.Javierre, B.Bugler, C.Arnould, V.Rocher, J.S.Iacovoni, A.Biernacka, M.Skrzypczak, K.Ginalski, M.Rowicka, P.Fraser and G.Legube. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology. 2017
- Roukos, T.C.Voss, C.K.Schmidt, S.Lee, D.Wangsa and T.Misteli. Spatial dynamics of chromosome translocations in living cells. Science. 2013.
- Neumaier, J.Swenson, C.Pham, A.Polyzos, A.T.Lo, P.Yang, J.Dyball, A.Asaithamby, D.J.Chen, M.J.Bissell, S.Thalhammer and S.V.Costes. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. PNAS. 2012
- R.Schrank, T.Aparicio, Y.Li, W.Chang, B.T.Chait, G.G.Gundersen, M.E.Gottesman and J.Gautier. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature. 2018.
doi: https://doi.org/10.1242/prelights.31439
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the molecular biology category:
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Defective BRCA1-mediated DNA end resection drives tandem duplication formation and FANCM synthetic lethality
Marta San Martin
preLists in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |