








Effective concentrations enforced by intrinsically disordered linkers are governed by polymer physics
Posted on: 1 April 2019 , updated on: 30 October 2019
Preprint posted on 14 March 2019
Article now published in PNAS at https://www.pnas.org/content/early/2019/10/25/1904813116.short?rss=1
Close encounters mediated by disorder – a FRET biosensor reveals how the properties of intrinsically disordered protein linkers determine their compaction
Selected by Tessa SinnigeCategories: biophysics
Introduction
Intrinsically disordered proteins (IDPs) play important roles e.g. as scaffolding and signalling proteins, performing functions for which their conformational flexibility is key (1). In comparison, the linkers in multidomain proteins may seem a trivial example of disordered regions, yet they have a profound effect on biomolecular interactions. When two protein domains are physically connected by a linker, the number of encounters increases by orders of magnitude, and so do the binding and reaction rates. If the linker is long enough to connect the domains without strain, the effective concentration of the linked domains no longer depends on the actual concentration of the components, but instead becomes a function of the architecture of the linker (2). Although the end-to-end length or hydrodynamic radius of a disordered chain can be described by polymer physics, it is not clear to what extent these laws apply to IDPs, and how the linker sequence influences compaction of the chain.
Results of the preprint
The authors of this preprint set out to design an experimental system to systematically study the effects of linker composition on effective concentrations, allowing the compaction of different linkers to be deduced. To this end, they designed a FRET biosensor (figure 1A in the preprint) where two fluorophores that form a FRET pair were fused to the N- and C- termini of MBD2 and p66α, two proteins that form a coiled coil complex. MBD2 and p66α were separated by a disordered linker consisting of glycine-serine repeats. A point mutation that weakens the interaction was introduced in the MBD2 domain in the construct, so that the wild-type peptide could be used as an efficient competitor to determine the interaction strength.
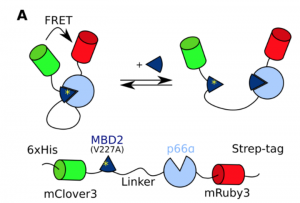
The authors performed titration experiments with the competing peptide at linker lengths ranging from 20 to 120 residues, and found that the curves shifted to a more open conformation at longer linker lengths. The effective concentration followed from the midpoint of the titrations, after correcting for the difference in affinity between the wild-type and the mutant MBD2 peptides. The authors could thus determine the effective concentration for each linker length, and found an exponential relationship ceff = 330 mM x N-1.46. In polymer physics, the end-to-end size of a chain (and equally the radius of hydration or gyration) scales with Nν, where N is the number of residues and ν is the scaling exponent that describes the compaction of the chain. In a three-dimensional volume, the effective concentration should accordingly scale with N-3ν, from which the authors could calculate that ν = 0.49 for the GS-linker. This corresponds to a slightly more compact conformation than previously reported for IDPs and denatured proteins, but this could be explained by the polar nature of the GS linker which allows for attractive backbone interactions.
Having established this system and demonstrated that the effective concentration can be used as a measure of linker compactness, the authors continued to systematically introduce different residue types to uncover the roles of charge, hydrophobicity and prolines. Charged residues are expected to extend polymer lengths because of repulsive interactions, and this is indeed what the authors found. The more charged residues were incorporated, the more the chains became extended. However, there was some variation between the different charged residue types. Whereas the scaling exponents of linkers with aspartate and glutamate residues were identical, lysine appeared to lead to more repulsion when introduced as a sufficiently large fraction, and arginine showed less repulsion. The authors suggest that arginine residues may compensate for electrostatic repulsion by their ability to engage in π-π stacking, which has also been suggested to account for the important role of arginine residues in liquid phase separation.
The introduction of proline residues gave less clear-cut results, and fluctuated with the fraction of residues incorporated in the GS linker. Proline increases the rigidity of polypeptide chains leading to more expansion, yet at a given fraction it may also induce folding into polyproline II helices. With regards to hydrophobicity, the incorporation of leucine residues did not have drastic effects, even though hydrophobicity should correlate with compaction of the chain. The authors note that this may play a bigger role in the context of more complex sequences such as native IDPs or unfolded proteins. Tyrosine, on the other hand, led to significant compaction of the linker which can be attributed to π-π stacking.
Another interesting case investigated in the preprint is that of polyampholytes, which contain both positive and negative charges. Incorporating equal fractions of glutamates and arginines into the linker led to a sharp increase in compactness above a certain threshold. Whereas one might expect that the combination of positive and negative charges alleviates charge repulsion, the effect that the authors found was much more striking: the scaling exponent reached the same value as that of globular, folded proteins, suggesting a similar level of compaction.
Finally, the authors examined whether or not the contributions from the different residue types were additive, which is highly relevant to understand the behaviour of the more complex sequences presented by native IDPs. When combining glutamate and proline residues, which both extend the chain, the authors found that the scaling exponent did not increase but instead was dominated by the glutamate residues. Furthermore, introducing hydrophobic leucine residues in the context of glutamate-containing linkers did not counteract the extension, altogether suggesting that chain compaction is governed by the strongest factor rather than being additive.
Why I chose this preprint
I had honestly never given much thought to linkers as IDP regions, and it struck me how the biophysical properties of disordered linkers have huge consequences for intramolecular interactions. The authors have taken a systematic approach to assess the relationship between sequence and linker compaction in this preprint that can be built upon in the future. The assay based on the FRET biosensor is very neat, and as indicated by the authors this approach can be extended to study the compaction of any IDP, which further increases the preprint’s contribution to the field.
Questions
Is there an estimate for the average linker length between folded domains in the human proteome? And how do the average charge and hydrophobicity of IDP linkers compare to the benchmark that was tested in this work?
I found it striking that the polyampholyte linker reached a scaling exponent similar to that of globular proteins. Does this mean that it actually folds, or would it be more like a molten globule state? How were the charged residues distributed? Would they, for example, be able to stabilise a helical structure by forming a regular pattern of ionic interactions?
The authors suggest in the discussion that disordered linkers may be involved in allosteric regulation, given that their compaction could be modulated by ligand binding or post-translational modifications, altering the effective concentration of the interacting domains. Are any such examples known?
In the case of posttranslational modifications, the current data suggest that the compaction of the linker is relatively robust and determined by the strongest contributor. In which scenarios (combination of residue types and post-translational modifications) would the authors envisage that large fluctuations could occur?
References
- Wright PE and Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16: 18–29
- Krishnamurthy VM, Semetey V, Bracher PJ, Shen N and Whitesides GM (2007) Dependence of Effective Molarity on Linker Length for an Intramolecular Protein−Ligand System. J. Am. Chem. Soc. 129: 1312–1320
doi: https://doi.org/10.1242/prelights.9695
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the biophysics category:
Mechanically-induced Septin Networks Protect Nuclear Integrity
Filipe Nunes Vicente
Loss of Sun2 ablates nuclear mechanosensing-driven extracellular matrix production and mitigates lung fibrosis
Beth Chopak
Shape independent fluidisation in epithelial monolayers
Sindhu Muthukrishnan
preLists in the biophysics category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
66th Biophysical Society Annual Meeting, 2022
Preprints presented at the 66th BPS Annual Meeting, Feb 19 - 23, 2022 (The below list is not exhaustive and the preprints are listed in no particular order.)
| List by | Soni Mohapatra |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Biophysical Society Meeting 2020
Some preprints presented at the Biophysical Society Meeting 2020 in San Diego, USA.
| List by | Tessa Sinnige |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Biomolecular NMR
Preprints related to the application and development of biomolecular NMR spectroscopy
| List by | Reid Alderson |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |