








Genome-wide maps of enhancer regulation connect risk variants to disease genes
Posted on: 30 September 2020
Preprint posted on 3 September 2020
Having a hard time finding the needle in the haystack? The activity-by-contact model connects risk variants and target genes to prioritize functional studies on GWAS.
Selected by Jesus Victorino*If you liked this preLights, please click on the thumb-up icon at the end of the page. Any comment, suggestion or question related to either scientific discussion or format will be more than welcome and very much appreciated. You can write directly at the bottom of this page or contact me by email or Twitter.
Last week I joined the #PreprintReviewChallenge, a great (and virtual) initiative organized by @ASAPbio_ and supported by @preLights, @PREreview_, @PeerCommunityIn & @PubPeer to build trust in #preprints. It was great to see more than 50 people, most of which were early-career researchers, gathering to chat about science, discuss about each other’s experience. With regard to science, I suggested the latest manuscript from the labs of Jesse Engreitz and Eric Lander. A week later (better late than never!) here’s my highlight including parts of the discussion that Iratxe Puebla, Julien Roux and myself had during the event.
I've just registered to participate in the @ASAPbio_ initiative #PreprintReviewChallenge to build trust in #preprints.@preLights @PREreview_ @PeerCommunityIn & @PubPeer joined to discuss and comment on recent science (no previous experience is needed)https://t.co/k5haJfujwy
— Jesús Mellamo (@JesusMellamoyo1) September 8, 2020
Summary & background
In the GWAS era that we live in, thousands of risk variants have already been associated to diseases [1]. For the most studied traits, the list of candidate loci contributing to common polygenic disorders is above a hundred and the number keeps growing as the sample size enlarges. The majority (>80%) of associated polymorphisms lie on the non-coding genome where they might affect the activity of regulatory elements and, therefore, gene expression [2]. But of which genes? And in which tissue?
Due to the huge number of possible scenarios for a given disease, it is of great importance to prioritize the candidate regions on which to focus functional studies. In this preprint, Nasser,
Bergman, Fulco, Guckelberger, Doughty et al. et al. build maps of enhancers with their target genes in over a hundred samples using a model of ‘Activity by contact’ where they take into
account chromatin accessibility, enhancer marks and enhancer-promoter interaction [3]. They integrate this data with variants associated to inflammatory bowel disease, among other traits,
and predict their target genes and tissue of relevance (Fig. 1). Using this approach, the authors identify an enhancer linked to inflammatory bowel disease that affects the metabolic state of mitochondria in immune cells. This work provides an interesting and powerful approach to characterize enhancer landscapes and their effect on the regulation of genes causing disease.
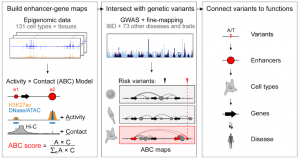
Figure 1. Activity by contact on over 100 biosamples to prioritized disease-associated pairs of enhancers-genes (taken from Fig 1a of the preprint).
Key results
– Mapping of over 6 million enhancer-gene connections across >100 biological samples.
– Prediction of the target genes for nearly 5,000 variants within enhancers across 72 traits.
– Prioritizing 14 new genes to inflammatory bowel disease, including PPIF.
– An enhancer controlling PPIF gene expression modulates mitochondrial function in immune cells responding to inflammatory stimuli.
How this work moves the field forward
In the last lustrum, GWAS have identified over a hundred associated variants to many common traits such as cardiovascular diseases [4, 5, 6, 7]. The significant increase in sample size of both cases and controls of such studies allowed many new SNPs to reach the widely accepted threshold for genome-wide significance, which is presumably going to keep growing as sample size keeps growing. In fact, several studies have also included sub-threshold SNPs when assessing the functional activity of associated non-coding regions since they are enriched for epigenetic signals specific to disease-relevant tissues [8]. However, they are very likely to be functionally weaker when compared to genome-wide significant variants, which is suggested by their lower contribution to genetic risk scores [6, 9].
Considering all this, I can’t help but wonder whether GWAS will identify variants forever or if we would reach a paradox situation in which every single variant in the genome will be associated to every single trait though in a very weak manner. Coming back to a more pragmatic view of the current situation, high-throughput screenings that functionally validate the activity of associated regions are going to be of seminal importance and, in this respect, massively-parallel reporter assay (MPRA) are very promising. Nevertheless, such assays have many limitations, such as their use exclusively in cell culture. Therefore, in order to elucidate the role of disease-relevant variants we still need genetic approaches of lower throughput that focuses on a reduced number of loci.
We could say that the two main limitations in the field are a constantly growing number of associations which not necessarily are relevant and the need of time-consuming techniques which are a bottleneck to fast-forwarding scientific discovery. For this matter, prioritization is of tremendous importance, since we cannot test everything thoroughly. Works like the one presented by Nasser et al. provide tools to find the needle in a haystack full of associations which will help dissect the regulatory code and understand the genetic contribution to disease.
Question to authors
1. In this preprint, the authors identified around 6 million candidate enhancers based on the ABC model in different biosamples. Do the authors know the estimated rate of false positive enhancers that should be expected among those? Are the authors planning on doing any sort of systematic validation to have a prediction of the performance of the predicting tool?
2. Each year, many new loci are identified for common diseases by GWAS. How easy would it be the systematic update of the new associations, in this case to inflammatory bowel disease, to include them in the priorized set of enhancer-genes?
3. In order to predict ABC enhancers the authors use data on chromatin accessibility, histone marks and HiC. How do the authors envision the inclusion of MPRA data to the ABC model in cell types where such resource is available?
References
1. GWAS catalog
doi: https://doi.org/10.1242/prelights.25020
Read preprint (1 votes)
(1 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genetics category:
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Also in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
preLists in the genetics category:
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSDB/GenSoc Spring Meeting 2024
A list of preprints highlighted at the British Society for Developmental Biology and Genetics Society joint Spring meeting 2024 at Warwick, UK.
| List by | Joyce Yu, Katherine Brown |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
2nd Conference of the Visegrád Group Society for Developmental Biology
Preprints from the 2nd Conference of the Visegrád Group Society for Developmental Biology (2-5 September, 2021, Szeged, Hungary)
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
Also in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |