








High-throughput functional analysis of lncRNA core promoters elucidates rules governing tissue-specificity
Posted on: 29 January 2019 , updated on: 30 January 2019
Preprint posted on 4 December 2018
What core promoter sequence features are important for gene activity? Mattioli et al. uncover an important role of overlapping motifs
Selected by Clarice HongCategories: genomics, systems biology
Background
Though transcription initiates from mRNA promoters, long non-coding RNA (lncRNA) promoters and enhancers (to produce enhancer RNAs, or eRNAs), each of these classes of genomic sequences have very different expression profiles. Specifically, lncRNAs and eRNAs are less active and more tissue-specific than mRNAs. The different expression patterns must be encoded by the genomic sequence itself, however, it remains unclear what sequence features determine different transcriptional patterns. Furthermore, a subclass of transcribed sequences, known as ‘divergent’ promoters, produce two stable transcripts in the sense and antisense direction respectively. Whether a ‘divergent’ transcript is produced by one promoter with unique sequence features or two proximal promoters remains unknown. Thus, to understand the sequence features underlying different promoter types, the authors used massively parallel reporter assays (MPRAs) to measure the intrinsic transcriptional activity of hundreds of promoters and enhancers in different cell types.
Key findings
The authors first grouped the genomic sequences that initiate transcription into 5 categories: eRNAs, intergenic lncRNAs (lincRNAs), divergent lncRNAs, mRNAs and divergent mRNAs. They then selected high-confidence transcription start sites (TSSs) for each category from 3 different cell lines (K562, HepG2 and HeLa) and designed sequences covering the core promoter to test for transcriptional activity. For the MPRA, each core promoter is linked to a unique barcode sequence that is transcribed. The activity of each promoter is then calculated by taking the RNA barcode counts divided by the DNA input barcode counts. Using this method, the authors found that both divergent mRNA and lncRNA promoters tended to be more active than their non-divergent counterparts, suggesting that divergent promoters are intrinsically stronger than non-divergent promoters. Furthermore, at least part of the tissue-specificity of core promoters appears to be encoded in the core promoter sequence itself, since the MPRA was able to recapitulate tissue-specific expression. Thus, the core promoter sequence alone can explain some of the differences between the different classes of promoters.
To determine the sequence features that discriminate between different promoters, the authors looked at two main features: the transcription factor (TF) motif architecture (the suite of TFs that binds to sequence) and the cell-type-specificity of the TFs that bind to the core promoter. TF motif architecture was further subdivided into two parts: number of independent binding sites in the sequence and the number of overlapping motifs. Using these three features, they fit a linear model to the MPRA data to see which feature contributes the most to core promoter activity. They found that while the number of binding sites and number of overlapping motifs (both under TF motif architecture) could explain some of the variation, cell-type-specificity of the TFs contributed almost nothing to core promoter activity. This suggests that the strength of a core promoter is dependent on its TF motif architecture, but this itself is not sufficient since they each only explain less than 20% of the variation.
Using the same metrics, the authors then looked at publicly available CAGE data (which measures the activity of each TSS in the genome) and found that overlapping TF motifs is correlated with higher core promoter activity and lower tissue-specificity. They thus hypothesised that disruptions in overlapping motifs would have a larger effect size than disruptions in individual motifs, since they are likely to have more severe consequences on promoter activity. To test this, they designed a second library of core promoter sequences from 21 disease-associated genes and 5 nearby lncRNAs and eRNAs with single nucleotide deletions spanning the core promoter. Indeed, the effect size of each deletion is somewhat correlated with the number of motifs it is predicted to disrupt, suggesting that overlapping TF motifs are indeed predictive of stronger promoter activity. This was also true for disease-associated single nucleotide polymorphisms (SNPs), as SNPs in overlapping motifs led to larger expression changes. From these results, the authors concluded that overlapping binding sites for different TFs allow a core promoter to be ubiquitously expressed across cell types and maintain high expression (Figure 1).
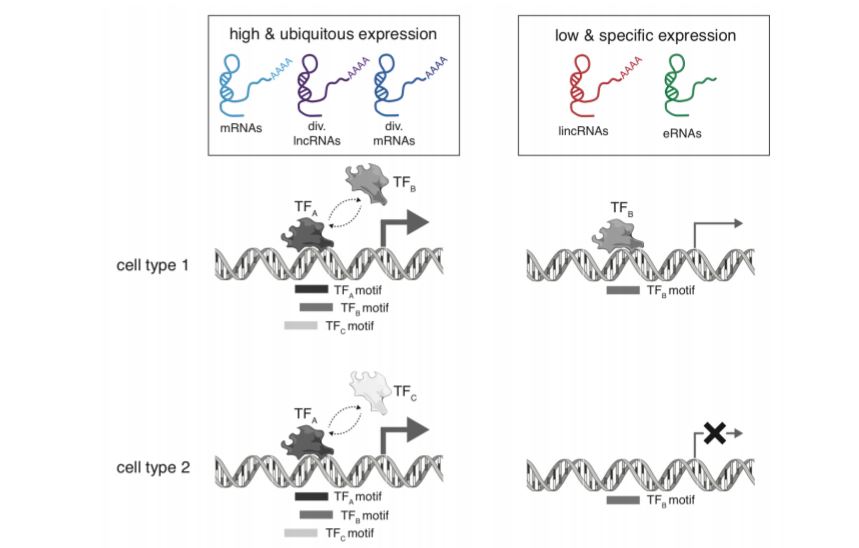
Figure 1: Summary of gene expression regulation by core promoters (Figure 5 from preprint). High and ubiquitous expression is associated with more overlapping TF motifs, while low and tissue-specific expression tends to have fewer TF motifs.
What I liked
As a student trying to understand the regulation of gene expression, the question of what sequence features of core promoters determine their activity is very interesting to me. This is especially exciting since we found out that so much more of the genome than we expected is transcribed. Since different groups of genes clearly have very different expression patterns, we need to find the rules governing these patterns. In this preprint, the authors took this one step further, and used some of the rules they learnt (overlapping TF motifs) to identify and determine the function of known SNPs in core promoters, which will be very useful for the understanding of non-coding disease variants. Furthermore, the MPRA is a powerful technique used to assay the activity of many DNA sequences, so I like that MPRAs are being used for this purpose. This also provides a great tool for the further study of TF binding sites and how variants affect TF binding and expression.
Future directions and questions
The biggest question that I have is what else is causing the differential expression levels and tissue specificity, since the features tested did not explain at least half of the variance. Can we consider other sequence features, for example, shape of the DNA? The specific combinations of TF motifs might also be important, since low affinity binding sites that are not usually picked up by motif finders can be used in the genome in combination with the right partners. Furthermore, are there any sequence features that might lead to a divergent vs non-divergent promoter? It also appears that the same rules used to explain the difference between categories of promoters can also be applied within each group of promoters, which suggests that perhaps things like TF motif architecture do not distinguish between the different promoter categories, but simply discriminates between high/low expression and tissue-specific expression. This begs the question of whether lncRNA and mRNA promoters and even eRNAs are categorically different, or whether they simply are transcribed according to the same rules to produce transcripts of different functions.
doi: https://doi.org/10.1242/prelights.8014
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Diversity and Spatial Segregation of TRP Channels in Choanoflagellates Provide Insight into the Evolutionary Origin of Animal Sensory Systems
Urvashi Goswami
BAF complexes maintain accessibility at stimulus-responsive chromatin and are required for transcriptional stimulus responses
Dina Kabbara
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Also in the systems biology category:
BAF complexes maintain accessibility at stimulus-responsive chromatin and are required for transcriptional stimulus responses
Dina Kabbara
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |