








Massively parallel protein-protein interaction measurement by sequencing (MP3-seq) enables rapid screening of protein heterodimers
Posted on: 7 April 2023 , updated on: 31 March 2025
Preprint posted on 9 February 2023
Article now published in Nature Chemical Biology at https://doi.org/10.1038/s41589-024-01718-x
High-throughput sequencing of protein-protein interactions quantifies more than 100,000 interactions simultaneously with interaction strengths spanning several orders of magnitude.
Selected by Benjamin Dominik MaierCategories: bioinformatics, genomics
Updated 31 March 2025 with a postLight by Benjamin Maier
Congratulations to Alexandr Baryshev, Alyssa La Fleur, and colleagues on their manuscript now published in Nature Chemical Biology! During revisions, they added a new section using AlphaFold2 and AlphaFold Multimer (v1–v3) to predict orthogonal binding (i.e. interactions) in an all-by-all screen. Initially, the sequence-based predictor iCipa showed stronger correlation with experimental log-fold change (LFC) values than any AlphaFold metric. However, by combining AlphaFold and Rosetta physics-based metrics, they trained regression models that performed comparably to iCipa, despite being trained on minimal data using less than 1/100th of the data used to train iCipa. When tested on new proteins outside the training set, AlphaFold Multimer’s accuracy declined, underscoring the continued necessity of high-throughput experimental assays for confirming orthogonality and assessing mutation effects on interactions.
Moreover, the authors tested three additional hypotheses adding supplementary figures illustrating their findings. First, they examined minimum read count filtering at various thresholds and found minimal effects on correlation with the dissociation constant (Kd), except for one series (NICP), where filtering improved Spearman’s rho (Supplementary Fig. 4). Second, they analysed weaker interactions by comparing enrichment and pseudoreplicate LFCs with Kd values for the five weakest interactions. They found that enrichment worked slightly better, suggesting that a non-DeSeq2 method might be better for weak PPIs (Supplementary Fig. 4c). Third, they screened intrinsically disordered PUMA peptides binding Mcl-1 at the BH3 pocket, confirming a strong correlation between MP3-seq and prior Kd measurements (Fig. 3h). Permutation tests showed no significant bias toward proteins with missing interaction data in DUET results (Supplementary Fig. 7c).
Background
Protein-protein interactions (PPIs) are ubiquitous in biological systems and play a crucial role in various biological processes such as cell proliferation, growth, and differentiation, as well as triggering downstream responses through cellular signalling (Westermarck et al., 2013). They are necessary to form complexes, transfer electrons (McMillan et al., 2013), regulate processes and transport molecules between different compartments. Proteins bind to each other through multiple mechanisms including hydrophobic bonding, van der Waals forces, and salt bridges (Xie et al., 2019). The binding specificity and strength (transient – permanent) varies greatly as there are promiscuous proteins unspecifically interacting with many other proteins as well as proteins that have exactly one partner. Hence there is a need for quantitative interaction measurements over several orders of magnitudes when performing proteome-wide studies.
As aberrant PPIs have been associated with numerous human diseases, including inter alia cancer, infectious diseases, and neurodegenerative diseases such as Alzheimer’s disease and Creutzfeldt–Jakob disease (Lu et al., 2020), targeting PPIs has become an important focus for novel treatments (Corbi-Verge et al., 2016 & Modell et al., 2016). However, most existing approaches for analysing PPIs are limited by scalability, a lack of knowledge of interaction sites, or the need for tedious laboratory work. Therefore, many existing methods have been improved and many new methods have been developed to better identify what proteins interact and how they interact.
In this study, Baryshev, La Fleur et al. (BioRxiv, 2023) developed a new Yeast Two-Hybrid approach with direct measurement of interaction strength by DNA sequencing (MP3-seq) for high-throughput study of protein-protein interactions.
Current Approaches to Study Protein-Protein Interactions
There are multiple high-throughput experimental and computational methods to characterise and quantity PPIs with yeast two-hybrid (Y2H) and tandem affinity purification-mass spectrometry (MS) being the most commonly used methods. Depending on the aim of a study, different factors (e.g. in vivo or in vitro, qualitative or quantitative approach, transient and/or stable PPIs, indirect or direct identification) should be considered when deciding on the method to address the research question. A more detailed overview of genetic, biochemical, biophysical and computational methods to study protein-protein interactions can be found below as supplementary comment. Additionally, I found the review from Stynen et al. (2012) and Rao et al. (2014) quite helpful even though they are not covering recent advances in the field.
Key Findings
New scalable Yeast Two-Hybrid approach (MP3-seq)
Baryshev, La Fleur et al. (BioRxiv, 2023) introduced a new scalable Yeast Two-Hybrid approach that can rapidly identify more than 100,000 pairwise protein-protein heterodimer interactions and quantify interaction strengths over several orders of magnitude. The workflow involves transforming all necessary DNA fragments into yeast and assembly in histidine auxotroph yeast. This is followed by positive plasmid selection after which the cells are split. While some cells are taken aside, the rest of the cells subjected to a His selection where cells can only grow when the proteins of interest interact, triggering the gene expression of an essential enzyme in the histidine biosynthesis pathway.
In more detail, yeast cells (histidine and tryptophan auxotroph) are transformed with all necessary components via electroporation. To reduce unwanted variability which could distort subsequent quantification, a centromere sequence is added as it ensures the expression of one pair of hybrid proteins per cell on average. As all elements can assemble to a plasmid in yeast through homologous recombination, no additional plasmid cloning (E. coli) or mating (yeast) step is required. Subsequently, the cells are grown in a selective media lacking Tryptophan (Trp), only allowing growth of yeast that have successfully transformed and assembled the plasmid (positive selection).
As the quantification method relies on comparing pre- and post-His-comparison, some cells were frozen, before the remaining cells were transferred to a selective media without histidine (His). If the two proteins to be screened interact with each other, the growth essential enzyme his3 is expressed allowing cells to grow in absence of histidine (His). his3 encodes the IGP dehydratase which catalyses an essential step in the histidine biosynthesis pathway. This means that cells that carry two strongly/permanently interacting proteins on their plasmid are expressing more IGP dehydratase allowing more histidine to be produced and thereby maximal growth. On the other hand, cells with transiently interacting proteins grow slower given the lower amount of available histidine.
Finally, cells from both pre- and post-His selection were lysed and the plasmid DNA extracted for subsequent amplification and sequencing. Based on the barcode counts before and after his-selection, one can compute the relative enrichment which serves as a proxy for the interaction strength.
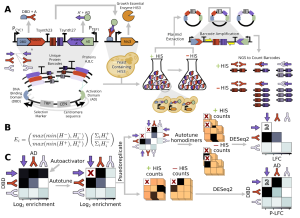
Development of a Computational Data Analysis Workflow
The authors have developed a computational data analysis workflow to analyse Mp3-seq data and quantify interaction strength (https://github.com/Seeliglab/MP3-DUET-AUTOTUNE). Following calculation of the enrichment, multiple corrections are performed including autoactivator removal, undetected PPI filtering, and accounting for experimental replicates.
In detail, the authors first computed the enrichment between the pre- and the post-His selection normalised by library size. Next, they removed proteins, which act as autoactivators meaning that they can non-specifically activate the expression of the reporter gene (here his3) in absence of a protein-protein interaction. They were identified by screening for proteins that displayed a high enrichment value for all interaction partners, which means that the his3 expression was unspecific. Subsequently, the authors screened for protein-protein combinations for which no barcode could be detected in the pre- and the post-His selection sequencing results and replaced them by the minimum value of detected pre-His reads. The corrected reads from both fusion orders (P1-DBD + P2-AD and P2-DBD + P1-AD, taken as pseudoreplicates) were then analysed by DESeq2 to determine the log2 fold changes which serves as proxy for the interaction strength.
Benchmark Results
The authors performed multiple benchmarking routines to make sure that their method is feasible for large-scale screening and that their results are in agreement with other state-of-the-art methods. These benchmarks included a large-scale assay of designed heterodimers, an approach with orthogonal coiled-coil dimers and an experiment with BCL family binders (control cell death primarily by direct binding interactions). Overall, their results demonstrated a good quantitative agreement with previously published results.
Perspective
Obtaining unbiased high-quality protein-protein interaction data is crucial for multiple fields including synthetic biology, drug development and molecular biology. Recent advances have paved the way for the design of novel drugs targeting PPIs resulting in multiple small molecules, peptides and antibodies currently being in clinical trials as PPI modulators. MP3-seq might be used as a complementary analysis to fragment-based drug discovery (FBDD) by building large-scale libraries of modular interaction domains.
Moreover, having more reliable experimental quantitative high-throughput protein-protein interaction data through MP3-seq as prior knowledge may allow for more sophisticated computational predictions of protein-protein interactions. I would assume that by combining it with AlphaFold-based docking methodologies like the protocol from Bryant et al. (2022) or AlphaFold-multimer which use optimised multiple sequence alignments, one could improve prediction results and filter out more false-positives further improving PPI predictions.
Finally, it would be interesting to see whether it is possible to tune the approach to specific cellular environments and other two-hybrid screening techniques, which would allow for context-specific and tailored analysis of protein-protein interactions.
What I liked about this preprint
What I really liked about this preprint is that the authors not only developed a very promising experimental method, but also a custom workflow to analyse their high-throughput Y2H data.
Collaborations between experimental and computational researchers are crucial in ensuring that the statistical and filtering analysis tools used downstream are specifically tailored to meet the demands of the experimental method. This leads to a more efficient, robust, and accurate interpretation of the results. Additionally, such collaborations foster innovation and interdisciplinary thinking, as both sides learn from and influence each other.
References
Bryant, P., Pozzati, G., & Elofsson, A. (2022). Improved prediction of protein-protein interactions using AlphaFold2. Nature Communications, 13(1). https://doi.org/10.1038/s41467-022-28865-w
Corbi-Verge, C., Kim, P.M. Motif mediated protein-protein interactions as drug targets. Cell Commun Signal 14, 8 (2016). https://doi.org/10.1186/s12964-016-0131-4
Lu, H., Zhou, Q., He, J. et al. (2020) Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Sig Transduct Target Ther 5, 213 . https://doi.org/10.1038/s41392-020-00315-3
McMillan DG, Marritt SJ, Firer-Sherwood MA, et al. (2013) Protein-protein interaction regulates the direction of catalysis and electron transfer in a redox enzyme complex. J Am Chem Soc., 135(28), 10550-10556. https://doi.org/10.1021/ja405072z
Modell, A. E., Blosser, S. L., & Arora, P. S. (2016). Systematic Targeting of Protein-Protein Interactions. Trends in pharmacological sciences, 37(8), 702–713. https://doi.org10.1016/j.tips.2016.05.008
Rao, V. S., Srinivas, K., Sujini, G. N., & Kumar, G. N. S. (2014). Protein-Protein Interaction Detection: Methods and Analysis. International Journal of Proteomics, 2014, 147648. https://doi.org/10.1155/2014/147648
Stynen Bram, Tournu Hélène, Tavernier Jan, & Van Dijck Patrick. (2012). Diversity in Genetic In Vivo Methods for Protein-Protein Interaction Studies: from the Yeast Two-Hybrid System to the Mammalian Split-Luciferase System. Microbiology and Molecular Biology Reviews, 76(2), 331–382. https://doi.org/10.1128/MMBR.05021-11
Westermarck, J., Ivaska, J., & Corthals, G. L. (2013). Identification of Protein Interactions Involved in Cellular Signaling. Molecular & Cellular Proteomics, 12(7), 1752–1763. https://doi.org/10.1074/mcp.R113.027771
Xie, N. Z., Du, Q. S., Li, J. X., & Huang, R. B. (2015). Exploring Strong Interactions in Proteins with Quantum Chemistry and Examples of Their Applications in Drug Design. PloS one, 10(9), e0137113. https://doi.org/10.1371/journal.pone.0137113
doi: https://doi.org/10.1242/prelights.34245
Read preprint (1 votes)
(1 votes) Have your say
Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the genomics category:
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
3 years
Benjamin Dominik Maier
Supplement: Current Approaches to Study Protein-Protein Interactions
*Genetic*
– Yeast Two-Hybrid (Y2H) System involves fusing one protein of interest to a DNA-binding domain (BDB) and another protein to an activation domain (AD) (Fields and Song, 1989). If the two proteins interact, the DNA-binding and activation domains come into close proximity, leading to the expression of a reporter gene. Newer versions of the assay (Weile et al., 2017; Luck et al. 2020) allow for screening of entire proteomes or random peptide libraries, which can enable unbiased identification of novel interactions.
– Protein fragment complementation assay (PCA) involves splitting a protein of interest into two fragments and fusing each fragment with a complementary protein fragment (Michnick et al., 2007). The resulting fusion proteins can only reconstitute the original protein activity if the two proteins interact and bring the two fragments together. This approach can be used to study protein-protein interactions in vivo and has been used for high-throughput screening to identify potential drug targets.
– Protein-array based methods involve immobilising large numbers of purified proteins on a solid surface (e.g. glass slide). The arrays can then be probed with (fluorescently) labelled proteins of interest to identify potential interaction partners. This approach enables the simultaneous screening of thousands of protein pairs, but it does necessitate laborious protein purification steps.
*Biophysical*
– Fluorescence/Bioluminescence Resonance Energy Transfer (FRET/BRET) involves labelling one protein with a donor fluorophore/luminescence and the other protein with an acceptor fluorophore (Sun et al., 2016). When the two proteins interact, the donor fluorophore transfers energy to the acceptor fluorophore, resulting in fluorescence, which can be measured by a detector.
– Mass spectrometry-based methods involve the fractionation of cell lysates by size or other physicochemical properties, followed by mass spectrometry analysis to identify co-eluting proteins (Richards et al., 2021). Although this approach can identify numerous potential interaction partners in an unbiased manner, it can be quite laborious since it requires protein purification steps.
*Biochemical*
– Immunoprecipitation-based methods such as Co-Immunoprecipitation (Co-IP) or Tandem Affinity Purification (TAP) involve the isolation of protein-protein interactions by adsorbing the protein complex onto beads using a combination of protein tags and specific antibodies (Lin & Lai, 2017), and subsequently identifying them by mass spectrometry or Western blotting.
– Surface Plasmon Resonance (SPR) involves immobilising one protein on a chip and flowing the other protein over it. The binding between the two proteins is measured in real-time based on the changes in the refractive index of the chip.
– Proximity-dependent labelling methods such as BioID and APEX enable identification of interacting proteins within a certain distance of the protein of interest and can be used to identify both stable and transient interactions in an unbiased way (Chen & Perrimon, 2017).
*Computational*
– Co-evolution Analysis makes inferences about protein-protein interactions using alignments (both sequence and structure) and phylogenetic distances. The approach can be used to predict functional residues and domains involved in the PPI.
– Molecular Docking Analysis uses structural templates of individual proteins to predict the structure of a complex. The approach is commonly used to screen large libraries of small molecule compounds for potential drug candidates that can target PPIs.
While most models require experimentally determined interactions as prior knowledge, there are also some approaches to predict interactions de novo.
Supplemental References
Chen, C. L., & Perrimon, N. (2017). Proximity‐dependent labeling methods for proteomic profiling in living cells. WIREs Developmental Biology, 6(4). https://doi.org/10.1002/wdev.272
Fields, S., & Song, O. (1989). A novel genetic system to detect protein-protein interactions. Nature, 340(6230), 245–246. https://doi.org/10.1038/340245a0
Lin, J. S., & Lai, E. M. (2017). Protein-Protein Interactions: Co-Immunoprecipitation. Methods in molecular biology (Clifton, N.J.), 1615, 211–219. https://doi.org/10.1007/978-1-4939-7033-9_17
Luck, K., Kim, DK., Lambourne, L. et al. A reference map of the human binary protein interactome. Nature 580, 402–408 (2020). https://doi.org/10.1038/s41586-020-2188-x
Michnick, S., Ear, P., Manderson, E. et al. (2007) Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov, 6, 569–582. https://doi.org/10.1038/nrd2311
Richards, A.L., Eckhardt, M. and Krogan, N.J. (2021) Mass spectrometry‐based protein–protein interaction networks for the study of human diseases, Molecular Systems Biology, 17(1). https://doi.org/10.15252/msb.20188792
Sun, S., Yang, X., Wang, Y., & Shen, X. (2016). In Vivo Analysis of Protein–Protein Interactions with Bioluminescence Resonance Energy Transfer (BRET): Progress and Prospects. International Journal of Molecular Sciences, 17(10), 1704. https://doi.org/10.3390/ijms17101704
Weile, J., Sun, S., Cote, A. G., Knapp, J., Verby, M., Mellor, J. C., … Roth, F. P. (2017). A framework for exhaustively mapping functional missense variants. Molecular Systems Biology, 13(12), 957. https://www.doi.org/10.15252/msb.20177908