








Mitotic heritability of DNA methylation at intermediately methylated sites is imprecise
Posted on: 24 March 2023 , updated on: 12 July 2023
Preprint posted on 1 February 2023
Probabilistic Inheritance of Intermediate DNA Methylation Sites Across Somatic Cell Division
Selected by Samantha Seah, Navin B Ramakrishna, Hui Ting ZhangCategories: genetics
Background
DNA methylation is an epigenetic mark that can play an important role in the regulation of gene expression, in particular during development and disease progression (Robertson, 2005; Smith and Meissner, 2013). It was previously thought that DNA methylation at CpG dinucleotides was precisely heritable through cell division (Cedar and Bergman, 2009). Recent works have indicated that such DNA methylation may be less stable and more dynamic than expected; in particular, intermediately methylated sites have been identified to spontaneously arise and have been observed to be inherited in an inconsistent manner (Arand et al., 2012; Pfeifer et al., 1990; Zhao et al., 2014). However, this phenomenon had not been studied systematically at a genome-wide level. A recent preprint by Hay and colleagues addresses this gap in knowledge by examining the fidelity of DNA methylation in 5% of CpGs in the mouse genome and characterising the intermediately methylated CpGs identified.
Key findings: Discovery and characterisation of the heritability of intermediately methylated sites in the genome
The authors established a system to study the heritability of methylation states across cell divisions, with the assumption that faithful methylation state inheritance results in only 0%, 50%, or 100% methylation at a site in a single cell, and thus also in a clonal population (Figure 1A, 1B). They immortalised mouse embryonic fibroblasts (MEFs) from two sibling C57BL/6J E13.5 mouse embryos, before sampling seven single cells from each line to establish a total of 14 clonal populations. Targeted DNA capture and bisulfite sequencing (tcBS-seq) was then used to profile around 1.2 million CpGs (encompassing approximately 5% of CpGs in the mouse genome). During initial quality control, aneuploidy at certain chromosomes was found in some or all the MEF-1 and MEF-2 lines. Chromosomes 12, 18 and 19 were therefore excluded from all downstream analysis, alongside the sex chromosomes, as MEF-1 was discovered to be male, and MEF-2, female.
66% of the CpG sites surveyed in the study were consistently fully methylated or unmethylated across all analysed cell lines (classified as M or U respectively by the authors) (Figure 1C, 2A, 2B). The authors computed a fidelity score for individual CpG sites based on the fraction of clonal lines that exhibited 0%, 50%, or 100% methylation at this site – indicative of faithful methylation state inheritance – and found that M and U CpG sites showed high fidelity scores, as expected. However, the authors also found that another 33% of the CpG sites were intermediately methylated in at least one set of the MEF-derived clonal cell populations. Of these, the majority showed intermediate methylation in one set of the clones and hyper/hypomethylation in the other (classified as MI and UI), while a minority was intermediately methylated in both sets of clones (classified as I). MI, I, and UI CpG sites were lower in fidelity score compared to M and U sites, were located in regions with lower CpG density compared to U sites, and exhibited lower similarity in methylation state with neighbouring sites (Figure 1D–F). They were also more commonly found in intergenic regions or in genes that showed low or no expression from matched RNA-seq data for each line (Figure 1G, 1H).
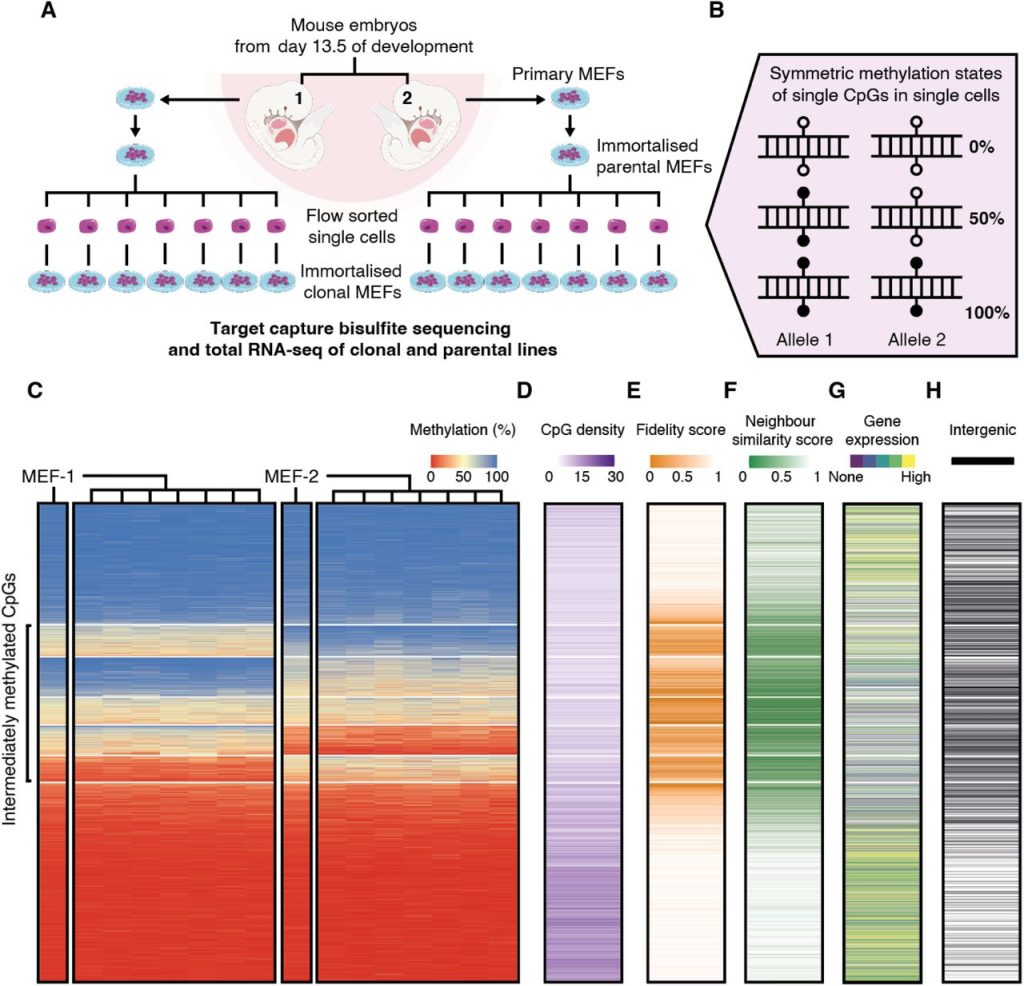
Figure 1. Figure 1 of Hay et al. (2023). Figure reproduced under a CC-BY-NC-ND 4.0 International License. (A) Schematic of the experimental design. (B) Definition of methylation states at a single-cell level. (C) Heatmaps of CpGs in parental and clonal lines, sorted by median methylation within k-means clusters. (D–G) Heatmaps of CpG density (D), fidelity score (E), neighbour similarity score (F), gene expression (G), and location (H) at these CpGs. Intergenic CpGs are represented by black lines in (H).
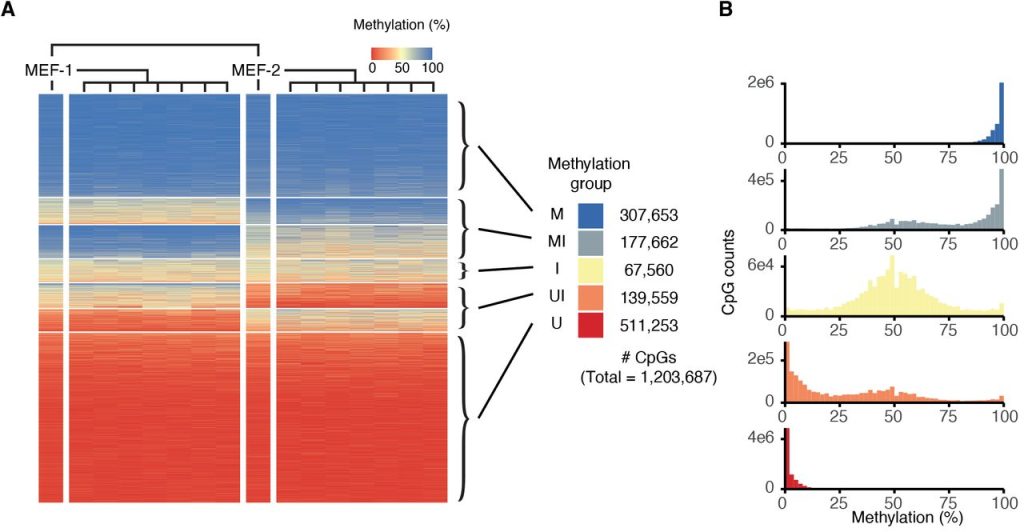
Figure 2. Panels A and B from Figure S3 of Hay et al. (2023). Figure reproduced under a CC-BY-NC-ND 4.0 International License. (A) Classification of CpGs into five classes: U = consistently hypomethylated across all the cell lines; UI = potential to be either hypo- or intermediately methylated; I = intermediately methylated; MI = potential to be either hyper- or intermediately methylated; M = consistently hypermethylated across all the cell lines. (B) Distribution of methylation levels in the five classes of CpGs.
Lower Methylation Fidelity at Lowly-Expressed Genes and Intergenic Regions
By analysing their RNA-seq data further, the authors were able to confirm known methylation differences over promoters and gene bodies – with promoters and the first exon of highly expressed genes typically hypomethylated, and the rest of the gene bodies typically hypermethylated (Suzuki and Bird, 2008). Importantly, they observed that these highly expressed genes had high fidelity scores, whereas genes with low or no expression tended to show intermediate methylation states throughout the gene structure, coupled with poor fidelity scores.
Hay and colleagues were also interested in the methylation dynamics and fidelity for CpGs over transposable elements (TEs), with knowledge that especially younger TEs may retain transcriptional potential and are thus more likely to be repressed by methylation. However, they found that CpGs within TEs in their dataset tended to be intermediately methylated, with no correlation in age with methylation levels and fidelity. Instead, they observed that methylation levels were more highly correlated with the genomic context of the TE: TEs in intergenic regions were more likely to have intermediate methylation levels and lower fidelity scores than TEs embedded in genic promoters (low methylation) or introns (higher methylation).
Linking Fidelity with Parental Methylation States
Beyond analysing clonal states, the authors proposed two models for the transmission of CpG states to analyse transmitted states between parental and clonal lines: faithful or stochastic inheritance (Figure 3A). The authors found that while CpG sites with high or low methylation levels faithfully recapitulated the parental methylation state, intermediately methylated sites did not. However, as these sites showed a modal and skewed distribution of methylation states in the clonal lines, they did not reflect a purely stochastic inheritance pattern either (Figure 3B). Thus, the authors concluded that methylation states at these CpG sites were inherited in a probabilistic manner — a combination of faithful and stochastic inheritance, with population heritability still observed. The authors then addressed the mechanism of this not strictly faithful methylation state inheritance by asking if this could be due to de novo methylation. An experiment using inducible knockouts of DNMT3A/3B in primary MEFs showed no change in global methylation levels, from which the authors concluded that inheritance of methylation states is likely reliant on the recently-discovered de novo functions of DNMT1 instead.
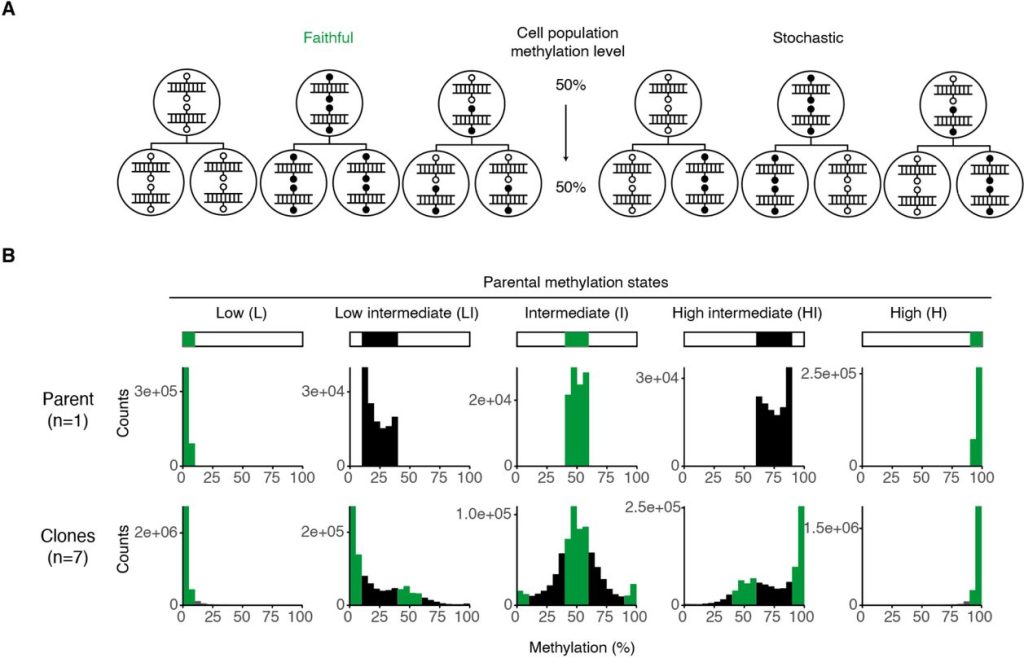
Figure 3. Panels A and B from Figure 3 of Hay et al. (2023). Figure reproduced under a CC-BY-NC-ND 4.0 International License. (A) Schematic depicting faithful and stochastic inheritance of methylation states across cell divisions. (B) Distribution of methylation states in the five classes of CpG sites in the clonal lines compared to their parent line (MEF-1).
In summary, the authors performed a global assessment of methylation states at CpG sites and their inheritance across cell divisions in somatic cells by comparing single-cell clonal populations with their parental cell lines. They identify and characterise a notable subset of these sites that are intermediately methylated, and which show a probabilistic inheritance of methylation states and an ability to both gain and lose methylation across cell divisions.
What we like about this work
This study represents attempts to formalise a clear and intuitive definition for fidelity of inheritance of methylation states through mitosis in somatic cells, with reasonable assumptions (symmetry of methylation state at a CpG dyad) for a first investigation. The authors approached the question methodically by constructing a well-controlled system to study this somatic epigenetic memory, developed using two parental lines and single cell clonal expansion to generate a total of seven subclones per parental line. In addition to their value in this study, these clonal cell lines (with matched parental lines) represent a rich data resource for in-depth or functional studies in the future.
The authors also go beyond simple characterisation of the intermediately methylated CpG sites. They noted that their fidelity scores do not include information about the parental state, making them insufficient to study inheritance of methylation states at intermediate sites with respect to the parental line. Thus, they then formalised two models of parental to daughter cell inheritance – faithful vs stochastic – and identified characteristics of each model as applied to a population-level dataset. This allowed them to observe that intermediate methylation states belong to neither model and to instead propose a probabilistic inheritance of methylation states that explains how intermediate sites can exhibit a combination of methylation states while maintaining population heritability.
The authors managed to rule out any de novo gain of methylation at the inheritance of these sites through inducible DNMT3A/3B knockouts, which led them to hypothesise that any gains may be due to de novo functions of DNMT1 instead. Testing this hypothesis directly is however understandably difficult as it involves separating the maintenance and de novo functions of DNMT1.
Finally, the findings from this study caution the use of CpGs with intermediate methylation states as biomarkers for disease and epigenetic age due to their imprecise inheritance.
Future work
A potential extension of this work could involve employing long-read sequencing options such as Oxford Nanopore Technology-seq to simultaneously sequence entire genomes and whole methylome data directly from non-amplified gDNA. This will not only enable the extension of the same analysis to all CpGs in the mouse genome, but will also facilitate the disentanglement of methylation data into 5mC and 5hmC, which BS-seq cannot differentiate. Moreover, by leveraging new Nanopore Duplex sequencing strategies that sequence both Watson and Crick strands, it may be possible to account for hemimethylation across a CpG dyad, which can also refine conclusions of methylation fidelity and transmission.
Another promising future exploration will be to examine single-cell DNA methylation states within the clonal lines (Clark et al., 2018). This will allow measurement of the precise mode of inheritance of various methylation states among the population of cells within a clonal line, without needing to infer these states from bulk-level datasets. Combining this with simultaneous scRNA-seq (Linker et al., 2019) can help monitor any fluctuations in expression, particularly at regions previously identified as intermediately methylated sites, to enhance our understanding of the significance of probabilistic inheritance. However, such an approach suffers from inadequate read coverage, which may limit the number of CpG sites that can be fully examined.
Additionally, one could explore functionally interrogating the significance of these intermediately methylated sites. This can be achieved through the targeted recruitment of DNA methylation writers or erasers fused to dCas9, to either introduce ectopic intermediate methylation sites and probe their inheritance, or to convert such sites to fully methylated or unmethylated sites.
Questions for the authors
- Do imprint control regions exemplify exceptional instances in the dataset, demonstrating high fidelity among clones and faithful parent-to-clone transmission, despite possessing intermediate methylation levels?
- Do the authors have any thoughts on the significance of the low-fidelity intermediately methylated CpG sites, considering their enrichment in intergenic regions and low-expressing genes? The authors have mentioned that the causal relationship, if one exists, between transcriptional activity and fidelity of inheritance is yet unclear. It is also intriguing to note that these intermediately methylated CpG sites are curiously heritable at the population level, despite being probabilistic at the clonal level.
- Do the authors suspect that the immortalisation process or sex differences could have influenced the methylation dynamics on the remnant autosomes included in the analysis of the MEF-1 and MEF-2 lines?
- The 5% of captured CpGs via SureSelectXT are enriched in certain regions of the genome, such as exomes and known DMRs, potentially at the expense of intergenic regions. With this in mind, would the authors be able to comment on how the findings might apply to the larger whole genome?
Further reading:
Shipony, Z., Mukamel, Z., Cohen, N. et al. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 513, 115–119 (2014). https://doi.org/10.1038/nature13458
Takahashi, Yuta, Mariana Morales Valencia, Yang Yu, Yasuo Ouchi, Kazuki Takahashi, Maxim Nikolaievich Shokhirev, Kathryn Lande, et al. Transgenerational Inheritance of Acquired Epigenetic Signatures at CpG Islands in Mice. Cell 186, 715-731.e19 (2023). https://doi.org/10.1016/j.cell.2022.12.047
Carlini V, Policarpi C, Hackett JA. Epigenetic inheritance is gated by naïve pluripotency and Dppa2. EMBO J. 41(7):e108677 (2022). https://doi.org/10.15252/embj.2021108677
Kazachenka, A. et al. Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-genetic Inheritance. Cell 175, 1259-1271.e13 (2018). https://doi.org/10.1016/j.cell.2018.09.043
Bertozzi, T.M., Becker, J.L., Blake, G.E.T. et al. Variably methylated retrotransposons are refractory to a range of environmental perturbations. Nat Genet 53, 1233–1242 (2021). https://doi.org/10.1038/s41588-021-00898-9
References
Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, Jenuwein T, Xu G, Leonhardt H, Wolf V, Walter J., In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet 8, e1002750 (2012). https://doi.org/10.1371/journal.pgen.1002750
Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009 May;10(5):295-304. https://doi.org/10.1038/nrg2540
Clark, S.J., Argelaguet, R., Kapourani, CA. et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun 9, 781 (2018). https://doi.org/10.1038/s41467-018-03149-4
P. Pfeifer, S. D. Steigerwald, R. S. Hansen, S. M. Gartler, A. D. Riggs, Polymerase chain reaction-aided genomic sequencing of an X chromosome-linked CpG island: methylation patterns suggest clonal inheritance, CpG site autonomy, and an explanation of activity state stability. Proc Natl Acad Sci U S A 87, 8252–8256 (1990). https://doi.org/10.1073/pnas.87.21.8252
D. Robertson, DNA methylation and human disease. Nat Rev Genet 6, 597–610 (2005). https://doi.org/10.1038/nrg1655
Linker, S.M., Urban, L., Clark, S.J. et al. Combined single-cell profiling of expression and DNA methylation reveals splicing regulation and heterogeneity. Genome Biol 20, 30 (2019). https://doi.org/10.1186/s13059-019-1644-0
D. Smith, A. Meissner, DNA methylation: roles in mammalian development. Nat Rev Genet 14, 204–220 (2013). https://doi.org/10.1038/nrg3354
M. Suzuki, A. Bird, DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9, 465-476 (2008). https://doi.org/10.1038/nrg2341
Zhao L, Sun MA, Li Z, Bai X, Yu M, Wang M, Liang L, Shao X, Arnovitz S, Wang Q, He C, Lu X, Chen J, Xie H., The dynamics of DNA methylation fidelity during mouse embryonic stem cell self-renewal and differentiation. Genome Res 24, 1296–1307 (2014). https://doi.org/10.1101/gr.163147.113
doi: https://doi.org/10.1242/prelights.34206
Read preprint (2 votes)
(2 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genetics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Site-Specific Inhibition of Translation Initiation via 2’-O-methylation
Leonie Brüne
preLists in the genetics category:
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSDB/GenSoc Spring Meeting 2024
A list of preprints highlighted at the British Society for Developmental Biology and Genetics Society joint Spring meeting 2024 at Warwick, UK.
| List by | Joyce Yu, Katherine Brown |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
2nd Conference of the Visegrád Group Society for Developmental Biology
Preprints from the 2nd Conference of the Visegrád Group Society for Developmental Biology (2-5 September, 2021, Szeged, Hungary)
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |