








RNase L reprograms translation by widespread mRNA turnover escaped by antiviral mRNAs
Posted on: 21 December 2018
Preprint posted on 4 December 2018
Article now published in Molecular Cell at http://dx.doi.org/10.1016/j.molcel.2019.07.029
Remodeling the transcriptome after viral infection – a mechanism of RNase L-mediated translational repression revealed
Selected by Connor RosenCategories: immunology, systems biology
Background:
The presence of foreign nucleic acids within a cell is a universal signal of pathogen invasion and the target for a wide range of immunity systems, ranging from restriction enzymes and CRISPR systems in bacteria to a coordinated array of cell-intrinsic and -extrinsic responses in mammals. Following detection of nucleic acids, typically derived from an infecting virus, by a range of dedicated pattern recognition receptors (PRRs), mammals initiate a cascade of signaling through the type I interferons to alert neighboring cells and induce a widespread anti-viral state. At the same time, infected cells induce cell-intrinsic defenses to limit viral replication and spread. One canonical response is the activation of protein kinase R (PKR) and RNase L to shut down cellular translation, preventing viral gene translation and subsequent viral particle production. However, it has been a mystery how the global shutdown of cellular translation and the requirement to continue to produce anti-viral proteins and type I interferons. This paper presents an elegant explanation solving this long-standing mystery in anti-viral responses.
Key findings:
- RNase L regulates stress granule assembly during an anti-viral response
The authors first demonstrated that stress granules, which are RNA-protein complexes formed downstream of PKR, are altered by RNase L activity. By using knockout cells and rescue experiments, the authors demonstrated that RNase L catalytic activity blocks classical stress granule formation at least partially through degradation of specific mRNAs important for stress granule assembly as well as nuclear translocation of a stress granule assembly factor PABPC1. Instead, cells with an active RNase L enzyme form smaller punctate foci the authors term RNase L-dependent bodies (RLBs).
- Anti-viral mRNAs escape RNase L-mediated degradation and continue to translate during an anti-viral response
The authors show widespread decrease in total mRNA levels during an anti-viral response, including a ~70% decrease in total poly(A)+ mRNA transcripts, including multiple transcripts that are not localized to stress granules. However, multiple anti-viral mRNAs, such as the cytokines IFN-b and IL-6, are not decreased. Whole transcriptome analysis revealed that the vast majority of mRNAs are degraded in an RNase L-dependent manner during an anti-viral response, but those that are not are highly enriched for anti-viral and IRF3-responsive genes. Importantly, IFN-b protein continued to be produced and secreted even after the anti-viral response began.
- There is widespread heterogeneity in individual cellular anti-viral responses
In the course of their microscopy experiments, the authors note that there is substantial heterogeneity between cells in their anti-viral response. For example, not all cells that induce RNase L activation produce IFN-b, and vice versa. The same is true for stress granule formation in RNase L knockout cells, as there is no strict connection between stress granule formation and IFN-b production.
Importance:
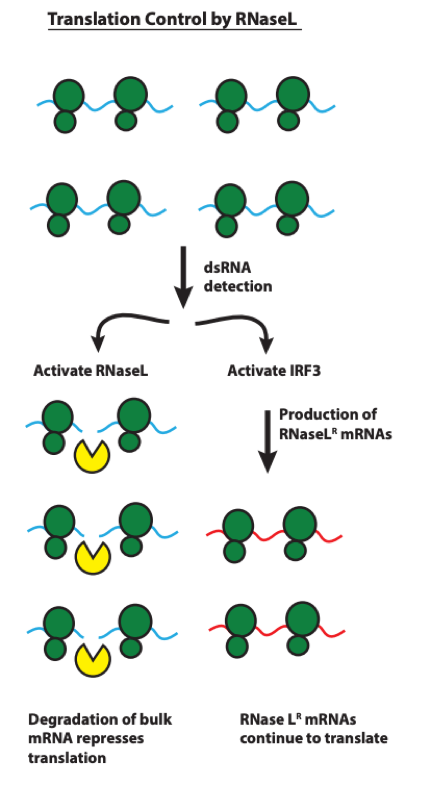
This preprint provides strong evidence for a model whereby RNase L activation, during an anti-viral response, leads to remodeling of the cellular transcriptome and translational control through escape from mRNA degradation of anti-viral mRNAs (see Figure 7, reproduced from the preprint below). This allows cells to accomplish the conflicting goals of restricting viral translation and initiating apoptosis while simultaneously producing anti-viral factors for cell-intrinsic defense as well as cell-cell communication. A long-standing mystery in anti-viral immunity is being resolved!
Figure 7. Model of RNase L reprogramming of translation via differential mRNA turnover.
Moving Forward:
- The system used throughout the paper is poly(I:C) transfection, which activates the dsRNA response. It will be very interesting to examine the cellular heterogeneity and RNase L-dependent responses in different viral infections, particularly in the setting of viruses with known or putative pathway antagonists. How have viruses evolved to subvert this defense system?
- The absence of a predictable sequence motif of RNase L-resistant mRNAs is very intriguing. The authors mention that many of the RNase L-resistant mRNAs contain AU-rich elements, which were very recently shown to be important for RNA localization to a membraneless organelle, the TIGER domain [Ma 2018]. It would be very interesting to see whether RNase L-resistant mRNAs are localized to the TIGER domain or a similar subcellular localization where RNase L may be excluded, providing a general mechanism for escape from degradation.
- There is so much exciting work that can emerge from the observations of heterogeneity in the anti-viral response between single cells. Is this heterogeneity a form of bet-hedging, to evade any single viral escape mechanism (e.g. by decoupling IFN-b production and stress granule assembly, cells can continue to produce interferon even in the presence of a viral PKR antagonist)? Is the total complement of anti-viral proteins produced similar between RNase L-active and inactive cells (or stress granule positive/negative cells), or is there some “specialization” leading to division of labor? How does communication between neighboring cells affect these decisions – the images seem to show neighboring cells taking on different “fates”, is this just from those images or is there paracrine feedback? Further microscopy, single-cell RNA-seq, and systems circuit modeling will all come to play – there’s so much to explore!
References:
- Ma, W. et al. A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3’UTR-Mediated Protein-Protein Interactions. Cell 175(6) 1492-1506.e19
doi: https://doi.org/10.1242/prelights.6701
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the immunology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
EBV reprograms autoreactive anti-CNS B cells as antigen presenting cells in multiple sclerosis
Léa Bastien et al.
Also in the systems biology category:
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
preLists in the immunology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |