








Single-cell gene regulatory network analysis reveals new melanoma cell states and transition trajectories during phenotype switching
Posted on: 13 September 2019
Preprint posted on 8 August 2019
This new preprint from the Aerts lab details how they used scRNA-seq to identify a new intermediate cell state during melanoma phenotype switching
Selected by Hannah BrunsdonCategories: bioinformatics, cancer biology
Background
Melanoma is the deadliest form of skin cancer. There is extreme genetic and cellular heterogeneity within each tumour as well as between patients. This, coupled with the ability of melanoma cells to switch between proliferative and invasive/drug resistant phenotypes means that identifying common cell states to target therapeutically is difficult. Despite this, two cell states have been found previously that recur across tumours and patients. One is the melanocyte-like state – marked by SOX10 and associated gene expression that controls the developmental melanocyte lineage program, and the other is the mesenchymal-like state – marked by SOX9 expression and associated with invasiveness and resistance to therapy.
In this preprint, Wouters, Kalender-Atak and colleagues aimed to investigate these states at the transcriptional level by performing single-cell RNA seq on nine diverse patient-derived melanoma cell lines, in order to identify gene regulatory networks (GRNs) common to different melanoma cell states.
Key findings
First, the team performed scRNA-seq on nine patient-derived 2D melanoma cultures (MM lines), as well as the A375 melanoma cell line. Each sample clustered together after dimensionality reduction, however there were also gene signatures in common across MM lines, chiefly the SOX10-high melanocyte-like state, and the SOX9-high mesenchymal-like state. Interestingly, some melanocyte-like MM lines also expressed mesenchymal- and immune-associated genes, representing an intermediate state. This can be visualised in the tSNE plot below:
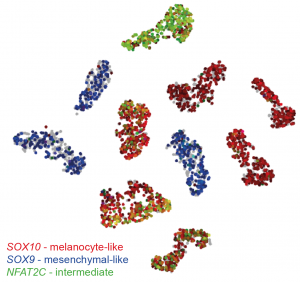
t-SNE plot of sc-RNA seq data generated using the online tool made by the authors (http://scope.aertslab.org/#/Wouters_Human_Melanoma). Each cell line forms its own cluster, however there are gene signatures in common. The SOX9 mesenchymal signature is restricted to three cell lines (blue). The other lines express SOX10, marking a melanotic phenotype (red). A subset of melanotic cell lines also express mesenchymal markers, as well as genes such as NFAT2C (green) which define a novel intermediate phenotype.
To investigate whether these transcriptional states translate to distinct tumour phenotypes, the authors conducted a single-cell migratory assay, whereby cells were seeded on a microfluidic chip containing channels for individual cells. Indeed, SOX9-expressing mesenchymal-like cells migrated fastest and the furthest along channels, whereas SOX10-expressing melanocyte-like cells migrated more slowly, with intermediate MM cells migrating somewhere between the two.
This intermediate cell state is not well understood in the literature, so the authors further investigated its transcriptional properties to identify an ‘intermediate cell state’ signature. SCENIC network inference was performed to predict which transcription factors and their targets – known as a regulon – governing each cell state. The intermediate cell states shared several regulons with both melanocyte-like and mesenchymal-like states, however some regulons including EGR3, RXRG and NFATC2 were specific to this state, suggesting this intermediate-state is a distinct cell identity. These identities were also found by Omni-ATAC-seq and in the TCGA database.
The authors next investigated a hypothesis that SOX10 disruption in would cause a transcriptional shift in melanocyte-like cells towards a more mesenchymal-like state. Indeed, bulk RNA-seq of intermediate-like cell lines at 24 hour intervals after siRNA-mediated knock-down of SOX10 showed progressive downregulation of melanocyte lineage markers, and upregulation of mesenchymal-like transcriptional signatures. By repeating SCENIC network inference along the trajectories pseudotime, the team could then order the key transcriptional events occurring after SOX10 knockdown. First, the cell cycle was paused due to inactivation of DNA polymerases and E2F/MYB transcription factors. Second, the melanocyte transcriptional program was inactivated by downregulation or pausing of melanocyte-associated transcriptional machinery. Lastly, genes associated with EMT were activated before induction of immune-related transcription factors.
Why I chose this preprint
Trying to identify clusters, common factors and tease patterns out of noisy-by-nature datasets can be quite a headache, so it is very interesting to see characterisation of common melanoma transcriptional signatures in what initially looks like very diverse samples. The authors have also made their scRNA analyses available at http://scope.aertslab.org/#/Wouters_Human_Melanoma, and it’s been fun to look at how my own genes of interest are expressed in the different cell lines. It will be interesting to see how targeting the intermediate cell state markers could change melanoma behaviour in a therapeutic context in terms of invasiveness and drug resistance.
Questions for the authors
Do you know if SOX9 knockdown or SOX10 overexpression in mesenchymal-like states would cause reprogramming towards a melanocyte-like state, and could this increase its drug sensitivity? Or are mesenchymal-like cells, or indeed melanotic cells towards the extremes of melanocyte-state more refractory to phenotype switching than intermediate-state cells?
Do you know if the established MM cell lines you used retain the degree of cellular and transcriptional heterogeneity that would be found if you were to obtain tumour cells directly from patients and compare these? Might the signatures you find be more enhanced by long term culturing in the same conditions?
How do you envision how the identification of these different states could help stratify treatments for patients, who might have different tumours at various places along the SOX10-melanotic/SOX9-mesenchymal spectrum?
doi: https://doi.org/10.1242/prelights.13744
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the cancer biology category:
Nature-Inspired Nanoparticle Adiposomes Enable Targeted Delivery of Hydrophobic Drug for Anti-Cancer Treatment
Elizabeth Pyman
Targeting CXADR-mediated AKT signaling suppresses tumorigenesis and enhances chemotherapy efficacy in Ewing sarcoma
TheLangeLab et al.
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |