








A lung-on-chip model reveals an essential role for alveolar epithelial cells in controlling bacterial growth during early tuberculosis
Posted on: 15 July 2020 , updated on: 16 July 2020
Preprint posted on 14 June 2020
Article now published in eLife at http://dx.doi.org/10.7554/eLife.59961
Surfactant secreted by alveolar cells protects from Mycobacterium tuberculosis
Selected by Snehal KadamCategories: bioengineering, microbiology
Context and background: Tuberculosis is a major healthcare burden, affecting a large proportion of the world (10 million individuals affected worldwide in 2018 (WHO)). A respiratory infection caused by Mycobacterium tuberculosis (Mtb), it begins with host-pathogen interactions at the air-liquid interface in the lung in early stages. Pulmonary surfactant, a mixture of lipids and proteins secreted by alveolar cells, has been traditionally known for its role in reducing surface tension and contributing to overall lung alveolar function [1]. Due to the fatality of surfactant deficiency and complexity of animal models, it has been difficult to probe the effects of surfactant on host-pathogen interactions. Organ-on-chip models combine the simplicity and feasibility of in vitro models with the ability to recapitulate physiologically relevant conditions like in in vivo models. This preprint looks at the role of surfactant released from alveolar epithelial cells using a model that recapitulates the air-liquid interface of lungs on a chip.
Experimental setup: The lung-on-chip model was made using polydimethylsiloxane (PDMS, a silicone-based organic polymer widely used for microfluidics) and had two chambers – air (alveolar) and liquid (vasculature) – separated by a porous membrane (see figure). The alveolar chamber contained alveolar epithelial cells (AECs), and the vascular chamber had endothelial cells. GFP-labelled macrophages were also added to the alveolar chamber, which could transverse the porous membrane to the endothelial side. Use of both AECs and macrophages allows one to study infection of both cell types. The chip was infected with fluorescently labelled Mycobacterium tuberculosis, WT, attenuated or avirulent mutants. Infection was visualized using time-lapse microscopy.
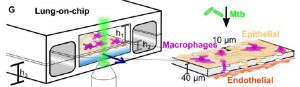
Important Results:
The lung-on-chip model recapitulates infection – This model makes use of two kinds of AECs – freshly isolated AECs which produce normal surfactant levels (NS), or in vitro passaged AECs (6-11 times) which have deficient surfactant levels (DS). These phenotypes were maintained in the chip, thus allowing the study of endogenous pulmonary surfactant. Addition of Mtb to the chip lead to both cell types getting infected.
Surfactant reduces growth rates of Mtb – The intracellular growth rate of Mtb post infection was monitored over five days. Though highly variable, a pattern emerged with respect to the surfactant conditions. NS conditions reduced growth rates of Mtb, with a larger proportion of cells showing very slow growth (non-growing fraction) when compared to DS conditions. This was evident in both AECs and macrophages. Exogenous addition of a surfactant solution in the DS condition leads to an increase in the non-growing fraction of cells, restoring control of Mtb growth. This indicates that surfactant does play a role in host-pathogen interactions, by protecting the host cells.
An attenuated strain, deficient for ESX-1 type III secretion system, is unable to grow irrespective of surfactant conditions, indicating that ESX-1 is still required in DS conditions. Similarly, another attenuated strain, previously shown to be unable to grow in lungs of mice, showed a similar phenotype in the AECs and macrophages of the chip model. This underscores the importance and ability of the lung-on-chip model to reproduce similar phenotypes as animal models.
Surfactant plays a role in attenuation of growth by removing virulence-associated lipids of Mtb –
Surfactant solution was seen to coat the bacteria upon exposure (but this effect was heterogenous). Additionally, sulfoglycolipids and trehalose dimycolate were partially removed from the surface of Mtb cells. These lipids are known to be important for virulence in inducing granulomas in mice and inhibiting cytokine production and immune response [2,3].
What I found interesting about this preprint:
The use of organ-on-chip approach enabled the study of endogenously secreted surfactant from alveolar cells, while maintaining the physiology and microenvironment of the air-liquid interface of lungs. Organ-on-chip models are gaining popularity in the field of infection research due to the ability to dissect host-microbe interactions at higher resolutions compared to animal models. I think this study makes a strong case for the use of such models, reproducing phenotypes and features known from animal models.
This study also shows that surfactant does more than its widely known role of reducing surface tension in the lungs, as it also protects the host again a pulmonary infection-causing pathogen. It would be interesting to see if such a protective role is seen against other pulmonary pathogens as well.
Questions for the authors:
- Where do you think the heterogeneity in coating of the Mtb surface with surfactant arises from? Does the correlate with any heterogeneity in the cell surface proteins of Mtb?
- Did you look at bacteria that cross over from the porous membrane into the endothelial side?
References/Further Reading:
[1] Torrelles, J. B., & Schlesinger, L. S. (2017). Integrating lung physiology, immunology, and tuberculosis. Trends in microbiology, 25(8), 688-697.
[2] Hunter, R. L., Olsen, M., Jagannath, C., & Actor, J. K. (2006). Trehalose 6, 6′-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. The American journal of pathology, 168(4), 1249-1261.
[3] Blanc, L., Gilleron, M., Prandi, J., Song, O. R., Jang, M. S., Gicquel, B., … & Vercellone, A. (2017). Mycobacterium tuberculosis inhibits human innate immune responses via the production of TLR2 antagonist glycolipids. Proceedings of the National Academy of Sciences, 114(42), 11205-11210.
doi: https://doi.org/10.1242/prelights.23098
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioengineering category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Also in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
preLists in the bioengineering category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Advances in microscopy
This preList highlights exciting unpublished preprint articles describing advances in microscopy with a focus on light-sheet microscopy.
| List by | Stephan Daetwyler |
Also in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |