








High-throughput aminoacyl-tRNA synthetase engineering for genetic code expansion in yeast
Posted on: 23 December 2021
Preprint posted on 3 December 2021
Article now published in ACS Synthetic Biology at http://dx.doi.org/10.1021/acssynbio.1c00626
Yeastward expansion: strategies by @heretharbtigerz and @Van_DeventerJim for tRNA synthetase engineering in S. cerevisiae to encode non-canonical amino acids
Selected by Zhang-He GohCategories: bioengineering, synthetic biology
Background of the preprint
The wide-ranging utility of proteins in therapeutic and scientific applications has driven demand for proteins to perform a greater range of functions. To that end, researchers have devised strategies to better modify proteins through improving post-translational modifications and chemical methods for selective protein functionalisation.
Another strategy to further expand the protein repertoire is to utilise non-canonical amino acids. There are 22 canonical amino acids which are found in nature (Figure 1), but researchers have expanded the genetic code to encode non-canonical amino acids into proteins. This can be achieved by loading host tRNA with non-canonical amino acids, with a ribosome subsequently transferring these amino acids from the tRNA onto the growing peptide chain. In these orthogonal translation systems, aminoacyl-tRNA synthetases load non-canonical amino acids onto tRNAs.
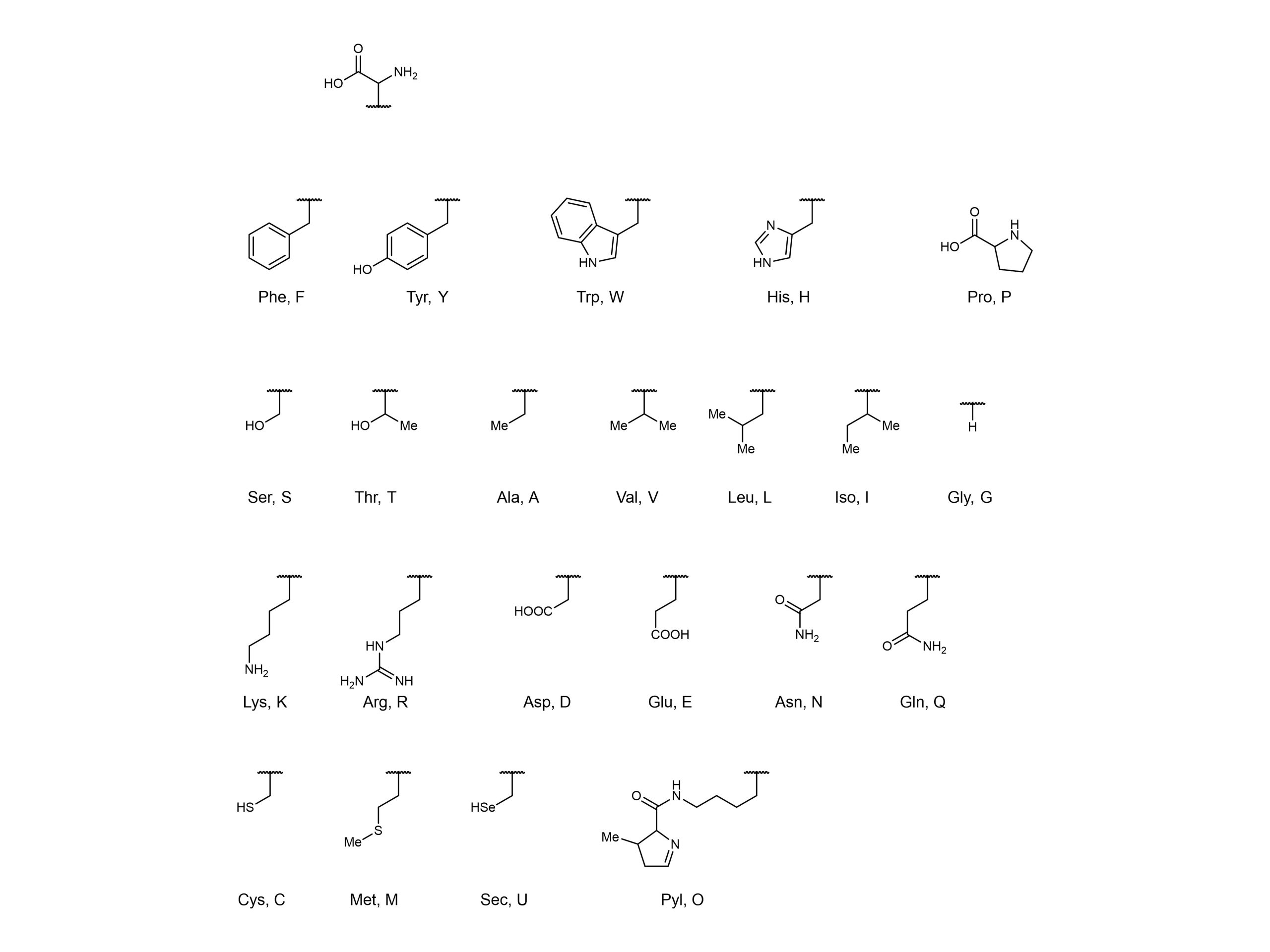
Figure 1. 22 canonical (naturally occurring) amino acids.
An important consideration with these methods is that they must not interfere with the host organism’s ability for protein synthesis. Thus, the challenge lies in the specificity of aminoacyl-tRNA synthetases in loading canonical amino acids onto their respective tRNAs. Modification of aminoacyl-tRNA synthetases commonly occurs through a multi-stage process. First, these aminoacyl-tRNA synthetases are taken from a model organism (such as E. coli). The residues in the aminoacylation site are then diversified, followed by multiple rounds of positive and negative selections to derive aminoacyl-tRNA synthetases that are specific for the target non-canonical amino acid.
However, the time-consuming nature of genetic code expansion necessitates the discovery of better methods to identify and evolve aminoacyl-tRNA synthetases for these purposes. The application of these strategies to other organisms would also be hugely beneficial.
Key findings of this preprint
In their preprint, Stieglitz and Van Deventer describe a flow cytometry-based, high-throughput screening platform tailored to genetic code expansion using aminoacyl-tRNA synthetases in yeast. The authors’ approach in their preprint can be classified into three broad categories: (A) library construction and screening, (B) implementation of different screening strategies, and (C) characterisation of the resultant derivative aminoacyl-tRNA synthetases.
(A) Library construction and screening
Stieglitz and Van Deventer first constructed saturation mutagenesis libraries of E. coli tyrosyl-tRNA synthetase and E. coli leucyl-tRNA synthetase; these two tRNA-synthetases were selected because their inclusion in yeast is less likely to disrupt native translation machinery.
The authors chose to mutate amino acid positions in these two aminoacyl-tRNA synthetases using saturation mutagenesis, thus substituting the codons at the selected positions with all possible amino acids. The authors reasoned that these changes would make aminoacyl-tRNA synthetases less specific for their cognate amino acids, in turn allowing them to better accommodate non-canonical amino acids which were bulkier or had longer side chains.
Using fluorescence-activated cell sorting (FACS), the authors screened both E. coli tyrosyl- and leucinyl-tRNA synthetases against various non-canonical aromatic and aliphatic amino acids selected to represent a range of structures and functions (Figure 2). Following multiple rounds of positive and negative selections, Stieglitz and Van Deventer identified several aminoacyl-tRNA transferases specific for non-canonical amino acids, some of which are new to yeast.
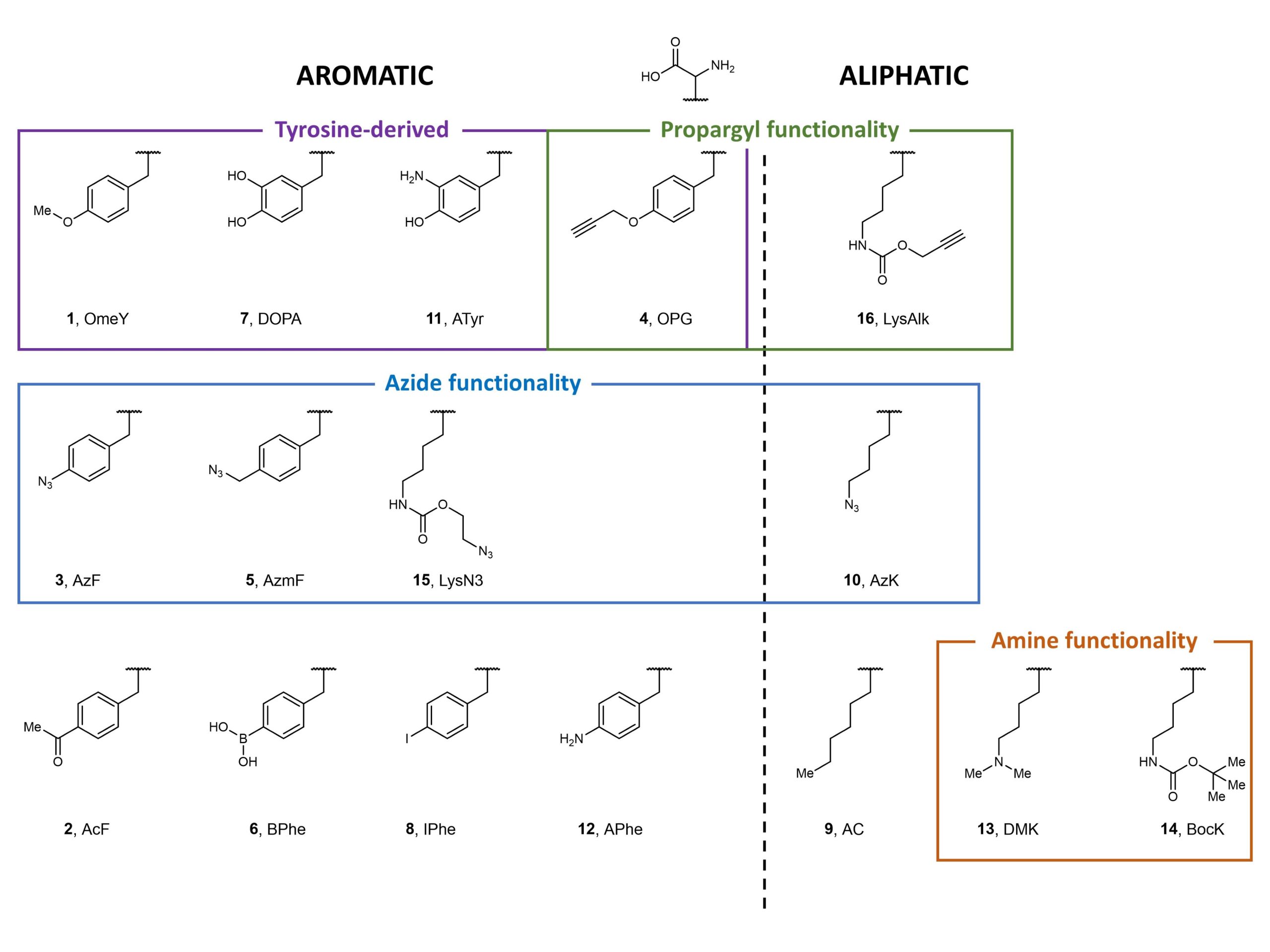
Figure 2. Non-canonical amino acids used to screen against E. coli tyrosyl- and leucinyl-tRNA synthetases.
Finally, the authors improved the activity of selected aminoacyl-tRNA synthetases using random mutagenesis through error-prone PCR—another new method in yeast. Through this work, the authors identified common mutations that were (a) directly adjacent to the active site or within the tRNA binding domain, (b) in the tRNA recognition domain, and, perhaps most interestingly, (c) at locations neither near the active site nor near the tRNA recognition sites. For this third category, the authors suggest that the observation that these mutations are present in multiple clones may imply that they are important in enhancing activity, and thus warrant further study.
(B) Implementation of modified screening strategies
Next, Stieglitz and Van Deventer aimed to investigate alternative induction conditions to isolate highly specific aminoacyl-tRNA transferases. This would overcome problems associated with the generation of polyspecific aminoacyl-tRNA transferases arising from the previously described methods. Using six structurally-similar aromatic non-canonical amino acids to optimise the screening process, the authors found that the use of specificity screening methods was essential in obtaining aminoacyl-tRNA transferases that were specific for a non-canonical amino acid compared to its structurally similar counterparts (in this case, OPG).
Simultaneously, the authors also explored the other side of the spectrum, aiming to enhance the polyspecificity of aminoacyl-tRNA synthetases via two “Tracks”. In Track 1, the authors used all 6 aromatic non-canonical amino acids (1 – 5, and 8) in positive sorts before a negative sort; in Track 2, just 1 aromatic non-canonical amino acid was used per positive sort. The authors concluded that Track 1 was easier to implement, but generally afforded less control. In contrast, Track 2 was more tedious, requiring monitoring after each sorting step, but allowed for better control of non-canonical amino acid incorporation.
(C) Characterisation of aminoacyl-tRNA synthetases
Finally, Stieglitz and Van Deventer evaluated the polyspecificity of their final enriched Track 1 library population against non-canonical amino acids 1 – 16 and analogues 17 – 22 (Figure 3). The authors found that several clones were able to encode the aliphatic non-canonical amino acid target, showing that their polyspecificity screens can generate aminoacyl-tRNA synthetases that can be used in conjunction with a breadth of non-canonical aromatic and aliphatic amino acids. In contrast to the aminoacyl-tRNA synthetases generated through these methods, whose specificity and polyspecificity were carefully tuned, the aminoacyl-tRNA synthetases generated from conventional methods exhibited variable levels of specificity and polyspecificity.
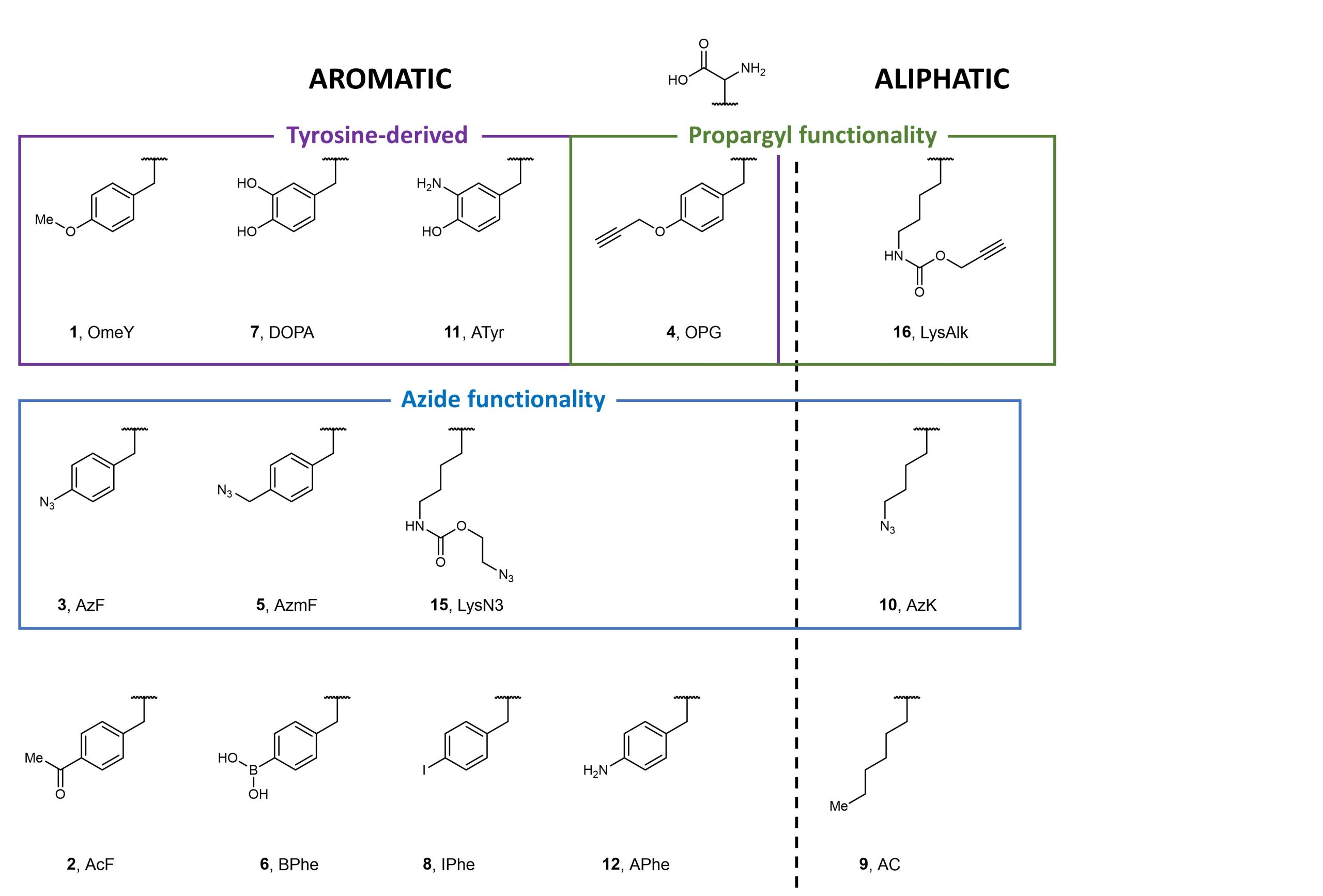
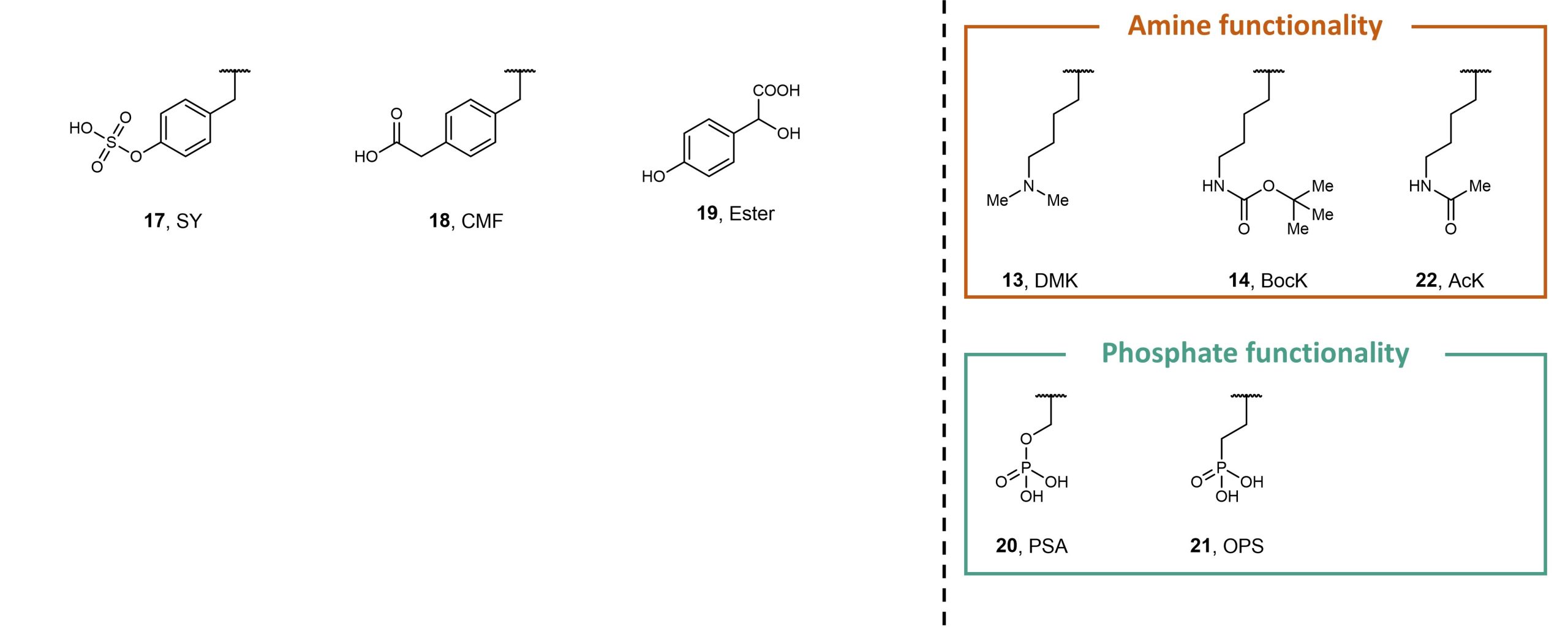
Figure 3. Non-canonical amino acids 1 – 18 and 20 – 22, and ester analogue 19.
What I like about this preprint
I chose to highlight this preprint because it overcomes important challenges in protein engineering. Gaining the ability to incorporate non-canonical amino acids into proteins reliably and robustly would be immensely useful to the scientific community for inventing new therapeutics and creating bespoke proteins to perform or probe specific cellular functions.
In their preprint, Stieglitz and Van Deventer have reported various strategies to finetune aminoacyl-tRNA synthetase specificity and polyspecificity. The ability to control these properties represents a significant advance over the usual methods, which do not afford such control. Crucially, the authors also demonstrated that these methods can also be used in yeast, a model organism other than E. coli. The authors also explored a broad scope of non-canonical aromatic and aliphatic amino acids containing functionalities that are orthogonal to subsequent protein functionalisation techniques, many of which I believe would also be useful handles for further protein functionalisation.
Future directions
Further work extending from this paper would likely involve an expansion of scope in two ways. The first involves the expanding the scope of the non-canonical amino acids that can be incorporated by these aminoacyl-tRNA transferases that the authors have generated and characterised. The second would be to identify (or generate) aminoacyl-tRNA transferases that would be able to incorporate this wider range of non-canonical amino acids.
In addition, the authors note that their work could inform subsequent studies on understanding sequence-activity relationships of these aminoacyl-tRNA transferases. This would not only aid in the abovementioned scope expansion, but would also improve the robustness of this technology in producing proteins more efficiently, that is, in greater yields and in higher purities.
Acknowledgements
Images created using Microsoft Powerpoint, ChemDraw, and BioRender.
Questions for authors
- Could you elaborate on how your methods compare to existing ones in terms of throughput? Specifically, how has throughput been increased via the use of flow cytometry?
doi: https://doi.org/10.1242/prelights.31204
Read preprint (1 votes)
(1 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioengineering category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Also in the synthetic biology category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Enzymatic bromination of native peptides for late-stage structural diversification via Suzuki-Miyaura coupling
Zhang-He Goh
Enhancer cooperativity can compensate for loss of activity over large genomic distances
Milan Antonovic
preLists in the bioengineering category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Advances in microscopy
This preList highlights exciting unpublished preprint articles describing advances in microscopy with a focus on light-sheet microscopy.
| List by | Stephan Daetwyler |
Also in the synthetic biology category:
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Advances in Drug Delivery
Advances in formulation technology or targeted delivery methods that describe or develop the distribution of small molecules or large macromolecules to specific parts of the body.
| List by | Zhang-He Goh |