








Multi-pass, single-molecule nanopore reading of long protein strands with single-amino acid sensitivity
Posted on: 4 December 2023 , updated on: 8 October 2024
Preprint posted on 20 October 2023
Article now published in Nature at https://www.nature.com/articles/s41586-024-07935-7#MOESM3
Next Generation Protein Sequencing: Achieving Single-Molecule Sensitivity and Full-Length Proteoform Identification!
Selected by Benjamin Dominik Maier, Samantha SeahCategories: bioengineering, molecular biology
Updated 8 October 2024 with a postLight by Benjamin Dominik Maier
Congratulations to Keisuke Motone, Daphne Kontogiorgos-Heintz and colleagues! Their manuscript has now been published in Nature (link to the journal article). The published manuscript has been highlighted in a Nature News Article by one of the reviewers and the full review history can be found here. Overall, the published manuscript does not differ much from the preprint. Notable additions are the detailled examination of the YY-dip signals to determine the step size of ClpX in their experiments, which was found to be approximately 1.96 residues per step. (see new Figures 2C-F). Moreover, a new section about monitoring and mapping enzymatic PTMs (see new Figure 5) has been added.
Background
Single-molecule protein sequencing technologies
Traditionally, proteins have been studied using Edman degradation (Edman et al., 1950), mass spectroscopy (MS) and enzyme-linked immunosorbent assay (ELISA) (Engvall and Perlmann, 1971). Edman degradation involves the selective removal of one amino acid at a time from the N-terminus of a protein and identifying it through a series of chemical reactions, while MS uses the specific mass-to-charge ratio of molecules for characterisation. ELISA relies on the specific binding between an antibody and its target antigen. Despite the age of these methods, we are experiencing a “renaissance” of these methods with recent improvements in both throughput and accuracy (e.g. Swaminathan et al., 2018).
Additionally, new methods relying on fluorescence and nanopores for single-molecule sensing have emerged in recent years. Yet, many of them are still under development and not widely applied. Single-molecule fluorescent protein fingerprinting allows the observation of individual protein molecules using DNA-PAINT imaging, offering insights into heterogeneity within samples. This method can track conformational changes, interactions, and structural dynamics at a granular level. Meanwhile, affinity-based approaches capitalise on specific molecular interactions to isolate and identify proteins or protein complexes. These newer techniques complement traditional methods, enriching our understanding of protein behaviour, structure-function relationships, and intricate cellular processes. An overview and description of existing and evolving methods can be found as in Figure 1 of Alfaro et al. (2021). For further details, we recommend Restrepo-Pérez et al. (2018) and Timp and Timp (2020).
Phosphoproteome Profiling
Protein phosphorylations are typically quantified through mass spectrometry-based methods (Yu & Veenstra, 2021) such as tandem mass spectroscopy. However, the readout of these methods does not achieve single-amino acid sensitivity, necessitating the use of computational algorithms and models to pinpoint the specific phosphorylation sites. To achieve high-throughput analysis, multiplexing methods have been developed recently (e.g. Mertins et al., 2018 using isobaric tags) to simultaneously measure multiple samples over time and various doses in a single assay minimising variability and noise between samples. More detailed information can be found for instance in Feng, Sanford et al. (2023), which we highlighted in a previous preLights article.
Key Findings
Overview
Keisuke Motone, Daphne Kontogiorgos-Heintz and colleagues have developed a new method to detect and sequence full-length proteins at single-amino acid resolution using a specialised protein unfoldase to guide proteins through a nanopore. By introducing a sequence that induces the unfoldase to slip, it is possible to re-read the protein molecules increasing sequencing accuracy. Moreover, the authors show that posttranslational modifications can be read out simultaneously to the amino acid composition. However, the authors also clearly state the limitations of their pioneering study and mention challenges to be solved in the future.
Key Finding I: Achieving single-amino acid sensitivity through the use of a ClpX unfoldase
There are similar challenges between the sequencing of nucleic acids and of peptides via nanopore – while chains can be translocated through nanopores with the use of a voltage difference across the membrane, such translocation is typically much too fast. Oxford Nanopore has resolved this problem by utilising a motor protein to ratchet the nucleic acids through the pore in a slow, controlled manner. Similarly, the authors have included a blocking domain within their protein, such that the protein gets stuck in the pore, before the ClpX motor is used to translocate the protein in a trans to cis direction at a regulated speed (Figure 1). This ensures that translocation takes place slowly enough for a usable current signal to be recorded.
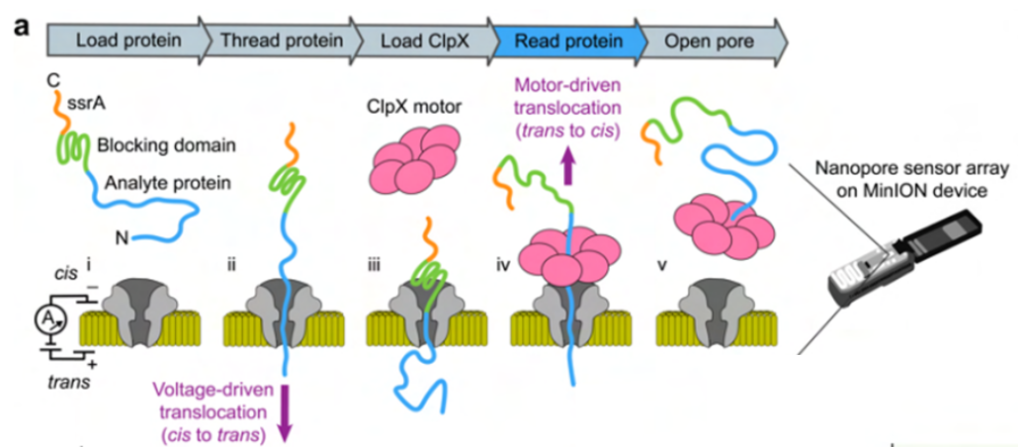
Fig. 1 Nanopore protein sequencing with the aid of an unfoldase. Figure adapted from Motone, Kontogiorgos-Heintz et al. (2023) Figure 1A, BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
The authors then designed 59 amino acid-long protein constructs that contain repeating sequence blocks and introduced single amino acid mutations at the central position within each block, with the ends of the blocks marked with double tyrosine mutations (which result in a characteristic dip in current) (Figure 2A). They were able to observe distinct and reproducible ionic signatures that corresponded to the single amino acid mutation within each construct (Figure 2B), showing that their method is sensitive to single amino acid residues, albeit in an artificial context.
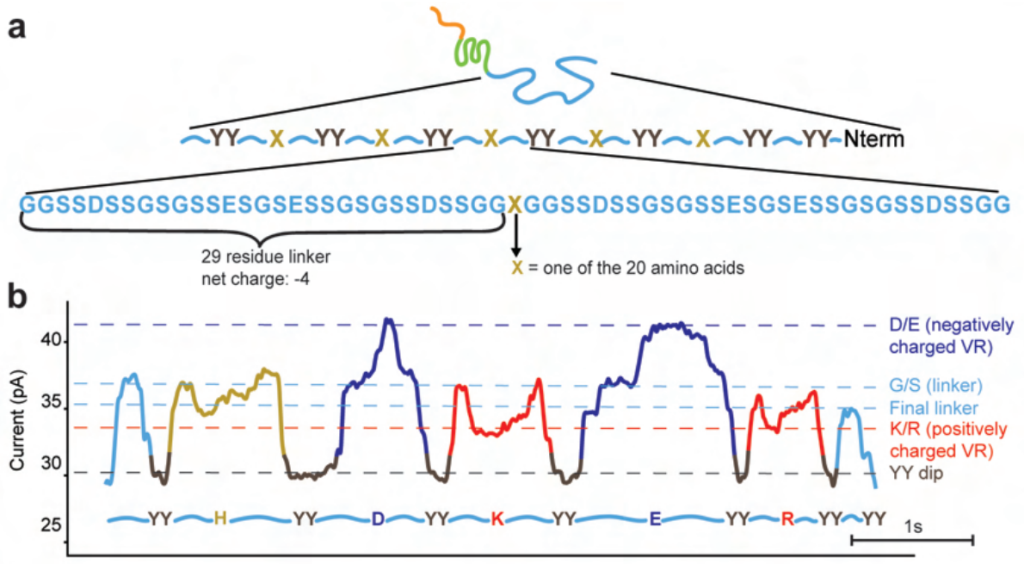
Fig. 2 Sequencing at single-amino acid level sensitivity . Figure adapted from Motone, Kontogiorgos-Heintz et al. (2023) Figure 2A and 2B, BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
The authors tested various machine learning models and were able to achieve an accuracy of 28% for all 20-way amino acid classifications (compared to 5.5% for a dummy model). They were also able to detect deamidation in asparagine, highlighting the potential of their technology to detect post-translational modifications.
Key Finding 2: Ability to re-read individual protein molecules
An important means of increasing the accuracy of sequencing methods is by resequencing the same molecule multiple times, and finding the consensus sequence. Such a method has been frequently utilised in long-read DNA sequencing, for example in UMIC-seq (Zurek et al., 2020) as utilised by Oxford Nanopore and in PacBio Circular Consensus Sequencing (Wenger et al., 2019).
The authors demonstrate that such a strategy is also possible with their nanopore-based peptide sequencing by including a slip sequence at the N-terminal part of the peptide, together with the blocking domain at the C-terminal part of the peptide. The blocking domain (as shown above) traps the protein in the pore to enable motor-binding and trans to cis motor-driven translocation, while the slip sequence causes the motor protein to slip, resulting in cis to trans (voltage-driven) translocation (Figure 3). This makes multiple rounds of trans to cis (motor driven) and cis to trans (voltage-driven) translocation possible, resulting in the sequencing of the same sequence multiple times.
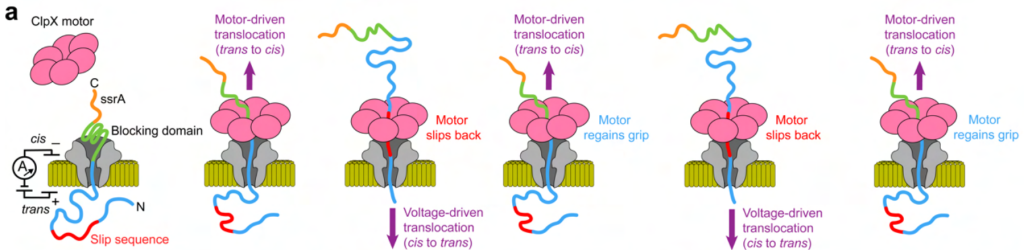
Fig. 3 Re-reading individual protein molecules. Figure adapted from Motone, Kontogiorgos-Heintz et al. (2023) Figure 5, BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Re-reading the same molecule improves the accuracy of the 20-way amino acid classification task from 28% (without re-reads) to 61% (with 10 re-reads). However, a shortcoming of this method is that the location at which the motor regains grip is not defined, such that some landmarks may be required to ascertain how the current traces align.
Key Finding 3: Ability to examine intact, folded protein domains
The end goal of a nanopore sequencing method would be to sequence natural proteins, many of which are folded or contain multiple folded domains. The authors tested their method on four different folded protein domains and could illustrate that the unfolding of folded domains is possible, although unsuccessful folding events can be seen, and that these are correlated with structural stability (Figure 4). This suggests that their method provides information on both structural (unfolding) and sequence (translocation) states of the proteins examined.
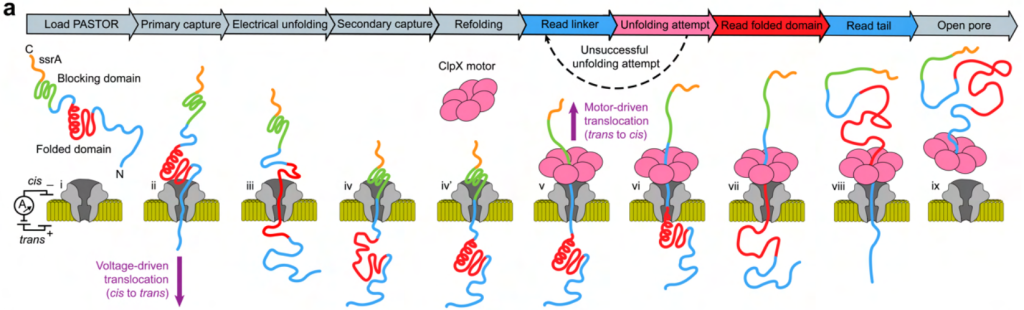
Fig. 4 Examining intact, folded protein domains. Figure adapted from Motone, Kontogiorgos-Heintz et al. (2023) Figure 6A, BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Conclusion and Perspective
We believe that the compatibility of the presented protein sequencing technology with existing ONT pores, which are already widely used for genome and transcriptome sequencing, makes this method a lot more accessible to scientists as their systems can be directly adapted for protein sequencing. However, given the early development stage of protein sequencing, it seems to us that the only real-world application right now is peptide barcoding, given that the single aa-level sensitivity can only be achieved in very synthetic and artificial contexts. Nonetheless, peptide barcoding could have exciting applications in protein-based screens, and the speed at which this field is developing continues to excite us.
Still, we found it quite striking that around 20 amino acids can occupy the CsgG sensing region when in a stretched conformation, which leads to 20^20 = 1.048576e+26 possible states. That’s a lot of different possibilities to be read out in a single signal, especially given that the change in this signal is in the order of 10−12 Ampere. The complexity of uncoding must really be quite high and should be close to/exceeding the physical and practical limits. We are keen to see how groups within the field will deal with this challenge as they work towards single amino-acid resolution de novo protein nanopore sequencing.
Questions to the Authors
Q1: Could the method be modified to simultaneously sequence proteins alongside other omics modalities? If so, what are the challenges that need to be tackled?
Q2: Can this method be combined with BOSS-RUNS (Weilguny, L., De Maio, N., Munro, R. et al., 2023) to disregard previously sequenced/uninteresting protein species in real-time and refocus on species that haven’t been covered (in as much depth) before?
Q3: How uniform is the motion through the pore using unfoldase-mediated translocation? And how important is it for protein sequencing that the motion is uniform?
Q4: Based on intuition, I would have guessed that a much smaller pore volume and a much thinner membrane would have been needed for protein sequencing given that amino acids are in the order of a few Ångströms (Å) in size compared to DNA (⌀ 2nm). Why is protein sequencing working with a pore optimised for DNA sequencing? And would a smaller pore and a thinner membrane improve readout accuracy?
Q5: Do you think the ClpX unfoldase will be able to unfold all protein domains or only a subset of domains? Do you have some insights regarding methods to aid protein unfolding that would be compatible with your set-up?
Q6: What are the next steps to get towards true de novo peptide sequencing with nanopores? And how long do you think it will take the field to get there?
Q7: Do you think that mass spectrometry will still be relevant in 20 years (once protein sequencing becomes reliable and widely-used)?
References
Alfaro, J.A., Bohländer, P., Dai, M. et al. (2021) The emerging landscape of single-molecule protein sequencing technologies. Nat Methods 18, 604–617. https://doi.org/10.1038/s41592-021-01143-1
Edman, P., Högfeldt, E., Sillén, L. G., & Kinell, P.-O. (1950). Method for Determination of the Amino Acid Sequence in Peptides. In Acta Chemica Scandinavica (Vol. 4, pp. 283–293). Danish Chemical Society. https://doi.org/10.3891/acta.chem.scand.04-0283
Engvall E, Perlmann P. (1971) Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. https://doi.org/10.1016/0019-2791(71)90454-x
Feng, S., Sanford, J. A., Weber, T., Hutchinson-Bunch, C. M., Dakup, P. P., Paurus, V. L., … Wiley, H. S. (2023). A Phosphoproteomics Data Resource for Systems-level Modeling of Kinase Signaling Networks. bioRxiv. https://doi.org/10.1101/2023.08.03.551714
Mertins, P., Tang, L.C., Krug, K. et al. (2018) Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat Protoc 13, 1632–1661. https://doi.org/10.1038/s41596-018-0006-9
Restrepo-Pérez, L., Joo, C. & Dekker, C. (2018) Paving the way to single-molecule protein sequencing. Nature Nanotech 13, 786–796. https://doi.org/10.1038/s41565-018-0236-6
Swaminathan, J., Boulgakov, A., Hernandez, E., Bardo, A. et al. (2018) Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat Biotechnol 36, 1076–1082. https://doi.org/10.1038/nbt.4278
Timp W, Timp G. (2020) Beyond mass spectrometry, the next step in proteomics. Sci Adv.;6(2):eaax8978. https://doi.org/10.1126/sciadv.aax8978
Weilguny, L., De Maio, N., Munro, R. et al. Dynamic, adaptive sampling during nanopore sequencing using Bayesian experimental design. Nat Biotechnol 41, 1018–1025. https://doi.org/10.1038/s41587-022-01580-z
Wenger, A.M., Peluso, P., Rowell, W.J. et al. (2019) Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37, 1155–1162. https://doi.org/10.1038/s41587-019-0217-9
Yu, L. R., & Veenstra, T. D. (2021). Characterization of Phosphorylated Proteins Using Mass Spectrometry. Current protein & peptide science, 22(2), 148–157. https://doi.org/10.2174/1389203721999201123200439
Zurek, P.J., Knyphausen, P., Neufeld, K. et al. (2020). UMI-linked consensus sequencing enables phylogenetic analysis of directed evolution. Nat Commun 11, 6023. https://doi.org/10.1038/s41467-020-19687-9
doi: https://doi.org/10.1242/prelights.36105
Read preprint (No Ratings Yet)
(No Ratings Yet)Have your say
Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioengineering category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Also in the molecular biology category:
Aging increases ovarian cancer growth, metastasis, and immunosuppression that can be alleviated by inhibiting hedgehog signaling
Zoha Sadaqat
A pre-rRNA positive feedback loop drives malignant ribosome biogenesis
Vaishali Grewal
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
preLists in the bioengineering category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Advances in microscopy
This preList highlights exciting unpublished preprint articles describing advances in microscopy with a focus on light-sheet microscopy.
| List by | Stephan Daetwyler |
Also in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
2 years
Benjamin Dominik Maier
The preprint has now also been highlighted in a Nature Technology Feature about the latest advances in Nanopore sequencing technologies: https://www.nature.com/articles/d41586-024-01280-5